Research Article
Authors:
Vignesh Sundararajan*1, Shuli Chen2, and Rhonda J. Rosengren2
1Department of Biotechnology, School of Bioengineering, SRM University, Kattankulathur, Chennai, India
2Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand
Corresponding author
Rhonda J. Rosengren, Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand, E-mail: rhonda.rosengren@otago.ac.nz
Received Date: 26 September 2017; Accepted Date: 28 October 2017; Published Date: 05 November 2017
Citation
Sundararajan V, Chen S and Rosengren RJ (2017) Raloxifene: Promises and challenges as a drug treatment for castrate resistant prostate cancer. Enliven: Toxicol Allied Clin Pharmacol 4(1): 001.
Copyright
@ 2017 Rhonda J. Rosengren. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, that permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.reproduction in any medium, provided the original author and source are credited.
Abstract
Prostate cancer is the most commonly diagnosed cancer in men. Although the cancers are initially androgen sensitive and respond to anti-hormonal therapy, over time they become refractory and grow in the absence of androgen. Such cancers, termed castrate resistant prostate cancer (CRPC), are aggressive in nature and have limited treatment options. Apart from androgens, estrogens also contribute to the initiation and progression of prostate cancer. Although estrogens are important for the normal development of the prostate gland, the estrogen receptors (ER) α and β are differentially expressed in tumors and thus offer a therapeutic target for the treatment of advanced, metastatic prostate cancer. Selective estrogen receptor modulators (SERMS) are a group of compounds that bind to ER and exert tissue specific agonist or antagonistic effects. Raloxifene, a SERM, approved for the treatment of osteoporosis in post-menopausal women, exhibits potent anti-cancer activity in in vitro and in vivo models of CRPC. However, poor bioavailability, extensive metabolism, and poor water solubility have reduced its efficacy in animal studies and clinical trials. With recent advances in nanotechnology, raloxifene has been successfully encapsulated in nanoparticles and exhibits superior pharmacokinetics than the free drug. Thus, this review has focused on the anti-cancer activity of raloxifene against CRPC, problems associated with the drug, results of clinical trials, and ways to improve raloxifene’s efficacy.
Role of Estrogen and Estrogen Receptors in the Normal Prostate
Although the development, differentiation, and functioning of the prostate gland are primarily mediated by androgen, estrogens also exerts profound direct and indirect effects on the prostate. The natural role of estrogens during prostatic development is uncertain, but excessive estrogenization during prostatic development can lead to benign prostatic hyperplasia (BPH) as well as prostate cancer in older males [1,2]. Investigating key developmental prostatic genes also showed that early exposure to high levels of estrogens initiated permanent structural and functional alterations to the prostate gland [3]. Estrogens can also have direct effects on the prostate gland in adults. It has been proposed that the growth of the stroma of the human prostate may be at least partly stimulated by estrogens which subsequently lead to an increase of 5α-reductase activity [4]. However, the above process results in an accumulation of dihydroxytestosterone (DHT), which in turn could over stimulate the growth of the epithelium. And the estradiol: DHT ratio increases massively within BPH tissue which directly implicates estradiol in the disease process [4]. The most important routes of indirect estrogen regulation are interference of androgen production by repression of the hypothalamic–pituitary–gonadal axis and direct effects on testis. One of the indirect mediators of estrogen effects on the prostate gland is stimulation prolactin release form the pituitary and some, but not all, of estrogen’s effects have been attributed to direct prolactin action on the prostate [5,6]. In addition, estrogens exert indirect effects on the inhibition of androgen production by negative feedback on the hypothalamic-hypophyseal-testicular axis, blocking lutinizing hormone secretion and testicular steroidogenesis of androgens [7]. Most of estrogen’s action in the prostate gland is mediated through two estrogen receptor (ER) subtypes ERα, expressed primarily in stromal cells, and ERβ, preferentially localized in the epithelium [8]. Both ERs are members of a large superfamily of proteins that function as ligand-activated transcription factors [9,10]. Due to their individual characteristics, ERα and ERβ have distinct biological functions. ERα was proposed to play a tumor suppressor role in the prostate gland and loss of its expression may be an early event in prostatic disease [11,12]. It has also been suggested that the action of ERα is not necessary for normal growth and function of the prostate gland [13,14]. However, it was observed that ERβ plays a central role in estrogen/antiestrogen signaling in normal and malignant human prostate endothelial cells [15]. Furthermore, ERβ has been proposed to have an anti-oxidant function and play an immunomodulatory role in the prostate gland [7].
Roles of ER in Prostate Cancer
While there is increasing evidence that ERs play a significant role in the growth and development of prostate cancer, its expression and function still remains controversial [16-22]. For example, the function of stromal ERα, remains largely unknown. Earlier studies suggested a possible role of ERα in promoting inflammation, proliferation, and metastasis [23,24]. However, another study has indicated that ERα in cancer associated fibroblasts (CAF) could promote prostate cancer cell proliferation and cell colony formation [25]. However, in a recent study of in vitro invasion assays and in vivo mouse models, it was observed that ERα could inhibit prostate cancer metastasis [18]. More recently, in vitro and in vivo studies on the role of stromal ERα in the later stages of prostate cancer progression showed that ERα in CAF was able to suppress prostate cancer cell invasion in the tumor microenvironment [19]. Due to this suppressing role, ERα levels in CAF was proposed to be utilized as a prognostic marker to predict cancer progression [19].
In comparison to ERα, the role of ERβ in prostate cancer is well studied. ERβ is considered to be tumor suppressor in prostate cancer, which makes it a promising drug target for the treatment and prevention of prostate cancer [26,27]. The anti-proliferative and pro-apoptotic role of ERβ in prostate cancer has been reported in both ERβ-knockout mice as well as human prostate tumors [28-30]. Additionally, 17β-estradiol and the ERβ-selective agonist diarylpropionitrile (DPN), but not the ERα-selective agonist propyl pyrazole triol (PPT), increased the incorporation of [3H]-thymidine and the expression of cyclin D2 in PC-3 prostate cancer cells, suggesting that ERβ mediates this proliferative effect [31]. It was also observed that ERβ also could cause apoptosis in Gleason grade 7 xenografted tissues as well as in androgen-independent PC-3 and DU-145 cell lines via caspase-8 [32]. Furthermore, with the identification of various isoforms of ERβ, different functions of ERβ in prostate carcinogenesis have been proposed. ERβ isoforms include ERβ2 to ERβ5 in which the most studied splice variants are ERβ2 and ERβ5. In one study, it was found that ERβ2 could increase prostate cancer cell invasion, while ERβ5 enhanced both cell migration and invasion [21]. In another study, however, evidence suggested ERβ2 was able to promote cancer cell migration and invasion in addition to cell proliferation, subsequently inducing the expression of factors involved in bone metastasis [16]. Thus, there is plenty of experimental evidence to suggest that targeting the ER is a viable therapeutic option in prostate cancer.
Castrate Resistant Prostate Cancer
Prostate cancer accounts for the largest number of diagnosed non-skin cancers in males and is the second leading cause of death amongst men in the United States [33]. A routinely used screening test for the detection of prostate cancer involves the measurement of serum levels of prostate specific antigen (PSA), where values greater than 2.5 ng/ml are considered positive for prostate cancer [34]. The common treatment for prostate cancer involves reduction of serum testosterone levels to <50 ng/dl via chemical or surgical castration [35]. Chemical castration often involves the use of androgen deprivation therapy medications such as gonadotrophin-releasing hormone (GnRH) agonists such as leuprolide, GnRH antagonists such as abarelix, adrenal ablating drugs such as ketoconazole, androgen receptor (AR) antagonists such as flutamide, and 5α-reductase inhibitors such as finasteride [36, 37]. While, most of the patients diagnosed with non-metastatic prostate cancer respond to initial treatments, the cancer can relapse into a form that does not respond to such treatments, producing distant metastasis [38,39]. The most common sites of prostate cancer metastases are bone, liver, lymph node, lungs, soft tissue, dura, and adrenal glands [40,41]. This aggressive and metastatic form of the disease is termed castrate resistant prostate cancer (CRPC) because the cancer cells grow in absence of androgen. Instead, there are various stimulatory signals that dominate such as tyrosine kinase receptors that are activated even when the level of circulating androgens are low [42].
For CRPC, the treatment options become limited and include the administration of narcotic analgesics, radiotherapy, cytotoxic chemotherapy, and use of palliative medications such as prednisone or hydrocortisone [43]. Although androgen deprivation therapy is useful in the management of advanced prostate cancer, it nonetheless has many side effects such as osteoporosis, sexual dysfunction, hot flashes, cardiovascular risk, and metabolic alterations [37,44]. Moreover, this form of advanced prostate cancer has poor survival rates, although recent studies have reported improvements over the international guideline estimate of ≤ 19 months [45,46]. Hence, there is a need for the development of safe and efficacious therapies without side effects for the treatment of CRPC.
Initially, docetaxel with prednisone was the common treatment strategy for patients with CRPC [43]. However, a number of drugs have been recently approved by the FDA for the treatment of CRPC, which includes enzalutamide (an androgen receptor inhibitor), abiraterone acetate (an androgen biosynthesis inhibitor), cabazitaxel (a microtubule inhibitor), sipuleucel-T (autologous cellular immunotherapy)[47], and radium 223 dichloride (an alpha-particle emitting radiotherapeutic drug)[48]. A number of studies reported the contribution of estrogen and estrogen signaling and androgen together with estrogen in the development of prostate cancer [49-53]. Moreover, it was reported that estrogen receptor α (ER) α was overexpressed in hormone refractory tumors and metastatic lesions in secondary sites such as lymph node and bone [54]. Thus, these studies provide a rationale to target the ER and its signaling in CRPC.
Selective Estrogen Receptor Modulators (SERMs)
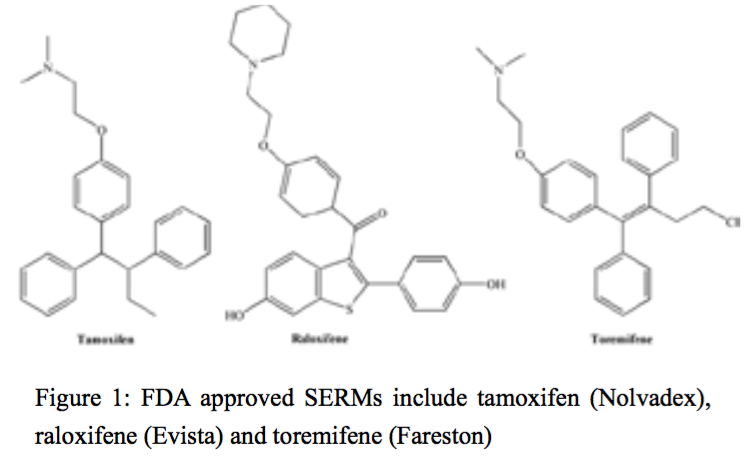
Selective estrogen receptor modulators (SERMs) are compounds that are able to bind to ERs in target organs and act as agonists or antagonists depending on the tissue. For example, they are often agonists in bone, liver, and the cardiovascular system, antagonists in brain and breast, and mixed agonists/antagonists in the uterus [55]. More than seventy different SERMs have been reviewed and they were subsequently classified into 5 different groups according to their chemical structure: triphenylethylenes, benzotiophenes, tetrahydronaphtylenes, indoles and benzopyrans [56-58]. The triphenylenes are planar, structurally rigid compounds which exit in either a cis- or trans- conformation. Benzotiophenes, such as, raloxifene contain a flexible carbonyl ‘hinge’ between the basic amine containing side chain and the olefin [59]. FDA approved SERMs include tamoxifen (Nolvadex), raloxifene (Evista) and toremifene (Fareston) (Figure 1).
Figure 1:FDA approved SERMs include tamoxifen (Nolvadex), raloxifene (Evista) and toremifene (Fareston)
Figure 1: FDA approved SERMs include tamoxifen (Nolvadex), raloxifene (Evista) and toremifene (Fareston)
Tamoxifen
Tamoxifen, a triphenylethylene derivative, acts as an ER antagonist in mammary tissue, but as an ER agonist in cholesterol metabolism, bone density regulation, and cell proliferation in the endometrium [60]. Tamoxifen is a first-generation SERM which has been used effectively for 40 years in the treatment of (ER)-positive breast cancer and for the prevention of breast cancer in high-risk women [61,62]. Tamoxifen has also been proposed as a treatment to prevent gynecomastia and/or breast pain, which is a very common side effect in men receiving antiandrogen/hormonal therapy for prostate cancer [63-66]. In general, gynecomastia results from an increase in the effective ratio of estrogens to androgens in breast tissue [67]. Up to 70% of patients receiving antiandrogen/hormonal therapy for prostate cancer have been reported to exhibit these side effects [64,65]. The side effects can have a strong negative impact on patient’s quality of life by causing physical pain and emotional discomfort, and are the major reasons for patients withdrawing from therapy [64,68,69].
Raloxifene
Raloxifene belongs to the benzothiophene group containing compounds and has been approved by the FDA for the treatment and prevention of postmenopausal osteoporosis as well as for the reduction in the risk of invasive breast cancer in postmenopausal women. Being an estrogen agonist in the skeletal and cardiovascular system, raloxifene is able to increase bone mineral density and decrease low-density lipoproteins. Raloxifene can also act as an antagonist on ERs in the breast and uterus to decrease the risk of cancer. Raloxifene is also under investigated for other potential indications, such as the primary and secondary prevention of cardiovascular disease in postmenopausal women and in breast cancer prevention in high-risk women [70,71]. Raloxifene binds to the ER with a similar Kd as 17β-estradiol (~50 pmol/l) [72]. Raloxifene exhibits rapid absorption and poor bioavailability as only 2% of an oral dose reaches the systemic circulation [73]. Raloxifene distributes extensively in the body, mainly to the liver, serum, lungs and kidneys [73] with the apparent volume of distribution is 2348 l/kg after a single oral dose (30 to 150 mg) [74]. Its low bioavailability results from extensive first-pass metabolism catalyzed by UDP-glucuronosyl-transferases (UGT) to form raloxifen-4'-glucoronide or raloxifene-6-glucoronide metabolites [75,76]. The majority of a dose of raloxifene is excreted primarily in the feces with less than 6% found in the urine [74].
In Vitro Effects of Raloxifene Against Prostate Cancer
Raloxifene has elicited cytotoxicity towards a variety of cancer cell lines including prostate cancer [77-82]. Compared to tamoxifen, raloxifene exhibited a higher affinity towards ERβ in U2OS bone cancer cells, MCF-7 breast cancer cells, Ishikawa endometrial cells, HeLa cells, and WAR-5 prostate cancer cells[83]. However, raloxifene also mediates its anti-cancer effect irrespective of androgen receptor (AR) status, and is effective in both androgen-sensitive and androgen-independent prostate cancer cells. Treatment of androgen-sensitive LNCaP and androgen-independent prostate cancer cell lines PC3, PC3M (ERα+/ERβ+) and DU145 (ERβ+) with raloxifene elicited significant cytotoxicity via the induction of apoptosis [84]. Piccolella et al., demonstrated that the anti-cancer effect of SERMs such as raloxifene was mediated via ERβ, in DU145 and PC3 cells that lack ERα. The aim of the study was to investigate if SERMs such as raloxifene and tamoxifen could mimic the anti-proliferative activity of a locally synthesized testosterone metabolite 5α-androstane-3β, 17β-diol (3β-adiol), which primarily exerted its effect via ERβ. Raloxifene and tamoxifen treatment (1 µM) for 48 h significantly decreased cell proliferation, migration and adhesion. Importantly, the above-mentioned effects were abolished in the presence of an ER antagonist ICI 182,780, which indicates that the effects of raloxifene were mediated via the ER[85]. Further studies have demonstrated that the anti-cancer effect of raloxifene was cell type-specific and also ERα/ERβ level-dependent. For example, in the androgen-dependent cell line EPN which expresses both ERα and ERβ, treatment with raloxifene caused cell cycle arrest in the G0/G1 phase, and apoptosis was induced as evidenced by increased expression of pro-apoptotic proteins caspase-3, Par-4 and downregulation of anti-apoptotic protein bcl-2. Cell proliferation in these cells was significantly reduced due to downregulation of c-myc and p27 mRNA expression [86]. Moreover, the expression of metallothionein II, an ERβ regulated gene, increased significantly after raloxifene treatment, indicating the ERβ-dependent role for raloxifene. On the contrary, in a stabilized epithelial cell line derived from a prostate cancer specimen (CPEC) that lacked ERα and expressed low levels of ERβ, only a weak apoptotic signal was observed [86].
These results therefore demonstrate that the anti-cancer activity of raloxifene in prostate cancer cells is mainly due to induction of apoptosis, cell cycle arrest, and decreased cell proliferation. However, the effect of raloxifene on the expression of AR remains ambiguous and also it was reported that raloxifene at high doses favored cell proliferation by mimicking the activity of androgens in a CPEC primary prostate cancer cell line expressing low level of ERβ and lacking ERα [86]. Hence, future studies should investigate the mechanism behind pro-androgenic effect of raloxifene at high doses, the effect of raloxifene in combination with other drugs, and also investigate the synthesis of analogs to further enhance the cytotoxicity of raloxifene towards CRPC.
In Vivo Effects of Raloxifene Against Prostate Cancer
Only a limited number of studies have investigated the anti-tumor effect of raloxifene in animal models. For example, Neubauer et al., investigated the anti-metastatic effect and overall survival in male Lobund-Wistar rats bearing the PAIII rat adenocarcinoma in the tail. Dosing of rats subcutaneously with raloxifene (20 mg/kg/day) for 30 days did not regress the primary tumor but it significantly decreased metastasis from the tail to gluteal lymph nodes (89%) and the lung (97%) [87]. Furthermore, PAIII rats that were dosed with raloxifene (40 mg/kg for 28 days) daily, exhibited significant increase in survival, compared to control. Raloxifene treatment also elicited a dose-dependent decrease (20%) in the ventral prostatic weight and 21% decrease in seminal vesical weight, and this was associated with a decrease in serum testosterone levels. An important finding from the study was that raloxifene mediated its anti-metastatic effect independent of the estrogen receptor as co-administration of estradiol benzoate (E2B) did not antagonize the effects of raloxifene, and the anti-metastatic activity of raloxifene did not involve any pharmacological interaction of raloxifene with E2B [87]. Lower doses of raloxifene have also shown tumor and metastasis suppression in an orthotopic model of CRPC. Specifically, raloxifene, when given orally (8.5 mg/kg/d, 42d) to male mice bearing PC3 tumors in their prostate, decreased tumor volume 70% and metastasis to renal lymph nodes 60% compared to vehicle control [88]. Interestingly, these results correlated with a 300% increase in the number of apoptotic cells in the tumor as well as an 84% decrease in ERα and a 92% decrease in ERβ as shown by immunohistochemistry of tumor slices. Thus, it has been shown that raloxifene administered orally can decrease metastasis in an orthotopic model. Decreasing metastasis is a critical drug action since it is metastasis, not the primary tumor that is responsible for the poor CRPC survival rate.
Raloxifene has also shown efficacy in transgenic mouse models. For example, Zeng et al., used a probasin/SV40 T antigen (Tag) transgenic mouse model, where mice develop adenocarcinoma of the prostate at the 15th week of age, to investigate the chemopreventive efficacy of raloxifene and nimesulide, a COX-2 inhibtor. The rats dosed with raloxifene (10 mg/kg/day) and nimesulide (400 ppm) exhibited a significant reduction in ventral prostatic weight. There was also a significant decrease in circulating testosterone levels in the group that received nimesulide plus raloxifene (10 mg/kg/d). Nimesulide on its own was ineffective in this model and the effects observed were due to the action of raloxifene. The rats dosed with raloxifene alone (5 mg/kg/d) or combined with nimesulide (400 ppm + 5 mg/kg/d or 10 mg/kg/d of raloxifene) exhibited downregulated androgen receptor levels in the ventral prostate. Moreover, raloxifene treatment at 10 mg/kg/d along with nimesulide significantly decreased cell proliferation as evidenced by decrease in the expression of proliferating cell nuclear antigen (PCAN) [89]. Investigation into the efficacy of raloxifene in androgen-dependent CWR22 and androgen-independent CWRSA9 prostate cancer xenograft models that express only ERβ, demonstrated that raloxifene elicited significant growth inhibition of tumors (64% for CWRSA9 and 68% for CWR22), although tumor regression was not observed. Raloxifene elicited its effect primarily by cell cycle arrest, as evidenced by the enhanced expression of G1 phase inhibitor, p27kip1[90].
Clinical Trials
Raloxifene has demonstrated promising results in the clinical trials. For example, a Phase II clinical study recruited 21 androgen-insensitive prostate cancer patients exhibiting disease progression after hormonal therapy. Patients were administered 60 mg oral raloxifene daily in 28 day cycles. 5 patients exhibited disease stabilization at the end of first cycle but only one patient stayed on the trial for 17 cycles [90]. Those removed were withdrawn due to increasing PSA levels, while two patients reported grade 3 toxicity. Raloxifene treatment also caused disease stabilization in pre-treated patients who exhibited disease progression prior to raloxifene treatment [90]. Raloxifene was also effective in inhibiting gonadotropin-releasing hormone (GnRH) agonist-induced bone loss in men with non-metastatic prostate cancer. 48 such patients who were already on a GnRH agonist for a minimum of 6 months were randomized and administered oral raloxifene at 60 mg/d for 12 months. Only 41 patients completed the study and an increase in bone mineral density of the hip was reported [91]. However, a phase 2 combination study involving the use of bicalutamide (50 mg) and raloxifene (60 mg) in 18 pre-treated men with progressive CRPC, for 6 cycles (28 days/cycle) demonstrated that raloxifene was safe without any grade 3 or 4 toxicity. In contrast, the combinatorial treatment did not elicit any significant clinical response [92]. It can be observed that although raloxifene was well tolerated and devoid of major toxicity, it elicited only limited clinical response in prostate cancer patients. However, future studies using raloxifene analogs or nanoformulation of raloxifene could be a strategy for identifying a drug formulation that has improved efficacy.
Raloxifene Analogs
Recent research has focussed on the development of synthetic raloxifene analogs in order to produce compounds that are more potent than raloxifene in their ability to antagonize ERα in a range of cancer cells. Though these analogues have not yet been tested in prostate cancer models, their improved efficacy and ER affinity make them ideal candidates for future studies in prostate cancer. For example, Shoda et al., synthesized novel raloxifene derivatives that acted as selective estrogen receptor destroyers (SERD), by acting as an inhibitor of ligand binding and destruction of the ER. The SERD activity of the derivatives was dependent on the length of the alkyl chains, and RC10, the most potent derivative, contained a decyl group on the amine moiety of raloxifene. RC10 exhibited ERα antagonistic activity via its proteosomal degradation in breast cancer cells and was superior to compound 18, an ER antagonist without SERD activity [93]. Other studies with Y134, a raloxifene analog with a piperazine side chain, showed that it was more potent than raloxifene as an ERα antagonist in CV-1 monkey kidney fibroblast cells co-transfected with ERα and ERβ [94]. Furthermore, it also inhibited estrogen-dependent proliferation of MCF-7 and T47D breast cancer cells.
Since there are studies that suggest selenium supplementation might protect against different cancers [95], selenium analogs of raloxifene have been synthesized. Results showed that the introduction of selenium remarkably increased the anti-proliferative activity [96]. The selenium analogs also exhibited less toxicity, compared to raloxifene. Substitution of hydroxyl groups with fluoro groups enhanced the cytotoxic profile of the selenium analogs. For example, the selenium analog 6a was cytotoxic towards a variety of cancer cell lines and also suppressed tumor growth 30% in a 4T1 breast cancer model when administered at 15 mg/kg. Interestingly, raloxifene was ineffective and was unable to inhibit tumor growth [96]. Other analogs synthesized include; organometallic analogs, radiolabeled analogues, constrained analogs that involved the use of tetracyclic coumarins, and thiacoumestans as scaffolds, as well as oxygen-, sulfur- and nitrogren-based analogs [97]. Thus, these results indicate that raloxifene analogs are promising drug candidates and are worthy of investigation in prostate cancer models.
Nanotechnology to Improve the Pharmacokinetics of Raloxifene
Although raloxifene is a promising drug candidate for the treatment of prostate cancer, it exhibits limited efficacy in in vivo models and clinical trials due to rapid absorption and pre-systemic clearance [73]. Nanoformulations of raloxifene have exhibited high loading capacity, reduced clearance and enhanced bioavailability. For example, poly (ε-caprolactone) nanocapsules of raloxifene had an encapsulation efficiency of >80%, a 2.1-fold increase in bioavailability, and sustained drug release when compared to the free drug [34]. Moreover, encapsulation of raloxifene in nanoparticles avoided first-pass metabolism, since the drug was up taken by M-cells of the Peyer’s patches in the intestine and further secreted into the lymphatic system [98]. Further studies to improve the bioavailability and efficacy of raloxifene reported that encapsulation of raloxifene in negatively charged nanoparticles/nanocapsules exhibited controled release of the drug and also elicited potent anti-proliferative effect against cancer cells [99,100]. Interestingly, raloxifene nanoparticles have been developed for increased bioavailability following both oral and intranasal administration [101-103].
Although raloxifene exhibited enhanced bioavailability and efficacy following encapsulation in nanoparticles, investigation into its efficacy against prostate cancer has been studied only recently. Taurin et al., investigated the cytotoxic potential of raloxifene encapsulated in styrene maleic acid (SMA) micelles (SMA-Ral) toward CRPC cells PC3 and DU145. Although SMA-Ral was cytotoxic toward both PC3 and DU145 cells at 5 and 10 µM, PC3 cells were more sensitive than DU145 cells in their response to SMA-Ral. Compared to the free drug, SMA-Ral was superior in eliciting not only cytotoxicity, but also increased apoptosis (11-fold), inhibited cell migration, invasion, as well as increased cell cycle arrest at the G0/G1 phase 20% [104]. The higher sensitivity of PC3 cells to SMA-Ral treatment was also shown via a 90% decrease in the expression of a splice variant of ER?, Δ5ER?, a decrease in the nuclear translocation of ERβ, and a 36% decrease in the expression of the EGFR expression and other downstream cell signaling proteins [104]. Confirmation of these results in xenograft models of CRPC using SMA-Ral demonstrated that the micelles accumulated to a greater degree in prostate tumors after 24 h, compared with the free drug [105]. Weekly administration of 1 mg/kg and 5 mg/kg of free raloxifene suppressed tumor growth 20% and 40%, respectively, after 4 weeks. However, SMA-Ral (1 mg/kg) was able to suppress tumor growth by 40% without any eliciting any toxic effects. This highlights the fact that encapsulation of raloxifene in nanomicelles was able to provide a similar tumor suppressive effect in vivo at a 5-fold lower dose [105]. Thus, raloxifene encapsulated into SMA micelles have potential to be developed into a safe an effective treatment for CRPC.
Conclusions
There is an urgent need to develop new targetable therapies to treat CRPC, a cancer with high metastatic ability and poor survival rates. Raloxifene, an FDA approved drug for the treatment of osteoporosis, has shown promising results in the studies conducted so far using in vitro, in vivo models and in clinical trials. Hence, it has the potential to be developed as an anti-cancer drug andcould be repurposed for use in the management of CRPC. However, the future use of this drug most likely lies within the field of nanomedicine due to its poor bioavailability and rapid pre-systemic clearance. Since raloxifene is a drug that is already in use, the development of a safe, less toxic and effective treatment for CRPC could become a reality in the near future for patients in desperate need of a novel targeted therapy against this advanced form of cancer.
References
- McPherson SJ, Wang H, Jones ME, Pedersen J, Iismaa TP, et al. (2001) Elevated Androgens and Prolactin in Aromatase-Deficient Mice Cause Enlargement, But not Malignancy, of the Prostate Gland. Endocrinology 142: 2458-2467.
- Rajfer J, Coffey DS (1978) Sex steroid imprinting of the immature prostate. Long-term effects. Invest Urol 16: 186-190.
- Prins GS, Huang L, Birch L, Pu Y (2006) The role of estrogens in normal and abnormal development of the prostate gland. Ann NY Acad Sci 1089: 1-13.
- Krieg M, Klötzl G, Kaufmann J, Voigt K (1981) Stroma of human benign prostatic hyperplasia: preferential tissue for androgen metabolism and oestrogen binding. Acta Endocrinol (Copenh) 96: 422-432.
- Gilleran JP, Putz O, DeJong M, DeJong S, Birch L, et al. (2003) The role of prolactin in the prostatic inflammatory response to neonatal estrogen. Endocrinology 144: 2046-2054.
- Lane KE, Leav I, Ziar J, Bridges RS, Rand WM, et al. (1997) Suppression of testosterone and estradiol-17beta-induced dysplasia in the dorsolateral prostate of Noble rats by bromocriptine. Carcinogenesis 18: 1505-1510.
- Prins GS, Korach KS (2008) The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 73: 233-244.
- Chang WY, Prins GS (1999) Estrogen receptor-ß: implications for the prostate gland. Prostate 40: 115-24.
- Katzenelienbogen JA, Katzenellenbogen BS (1996) Nuclear hormone receptors: ligand-activated regulators of transcription and diverse cell responses. Chem Biol 3: 529-536.
- George G.J.M. Kuiper, Mats Carlquist, Jan-Ake Gustafsson (1998) Estrogen is a male and female hormone. Science and Medicine 5: 36-45.
- Hernández J, Balic I, Johnson-Pais TL, Higgins BA, Torkko KC, et al. (2006) Association between an eestrogen receptor alpha gene polymorphism and the risk of prostate cancer in black men. J Urol 175: 523-527.
- Tanaka Y, Sasaki M, Kaneuchi M, Shiina H, Igawa M, et al. (2003) Polymorphisms of estrogen receptor alpha in prostate cancer. Mol Carcinog 37: 202-208.
- Eddy EM, Washburn TF, Bunch DO, Goulding EH, Gladen BC, et al. (1996) Targeted disruption of the estrogen receptor gene in male mice causes alteration of spermatogenesis and infertility. Endocrinology 137: 4796-4805.
- Donaldson KM, Tong SY, Washburn T, Lubahn DB, Eddy EM, et al. (1996) Morphometric study of the gubernaculum in male estrogen receptor mutant mice. J Androl 17: 91-95.
- Lau KM, LaSpina M, Long J, Ho SM (2000) Expression of estrogen receptor (ER)-α and ER-β in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res 60: 3175-3182.
- Dey P, Jonsson P, Hartman J, Williams C, Ström A, et al. (2012) Estrogen receptors β1 and β2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol Endocrinol 26: 1991-2003.
- Royuela M, De Miguel MP, Bethencourt FR, Sanchez-Chapado M, Fraile B, et al. (2001) Estrogen receptors alpha and beta in the normal, hyperplastic and carcinomatous human prostate. J Endocrinol 168: 447-454.
- Slavin S, Yeh CR, Da J, Yu S, Miyamoto H, et al. (2014) Estrogen receptor α in cancer-associated fibroblasts suppresses prostate cancer invasion via modulation of thrombospondin 2 and matrix metalloproteinase 3. Carcinogenesis 35: 1301-9.
- Yeh CR, Slavin S, Da J, Hsu I, Luo J, et al. (2016) Estrogen receptor α in cancer associated fibroblasts suppresses prostate cancer invasion via reducing CCL5, IL6 and macrophage infiltration in the tumor microenvironment. Mol Cancer 20:15-7.
- Celhay O, Yacoub M, Irani J, Dore B, Cussenot O, et al. (2010) Expression of estrogen related proteins in hormone refractory prostate cancer: association with tumor progression. J Urol 184: 2172-2178.
- Leung YK, Lam HM, Wu S, Song D, Levin L, et al. (2010) Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr Relat Cancer 17: 675-689.
- Zellweger T, Stürm S, Rey S, Zlobec I, Gsponer JR, et al. (2013) Estrogen receptor β expression and androgen receptor phosphorylation correlate with a poor clinical outcome in hormone-naive prostate cancer and are elevated in castration-resistant disease. Endocr Relat Cancer 20: 403-413.
- Bonkhoff H, Fixemer, T, Hunsicker I, Remberger K (1999) Estrogen receptor expression in prostate cancer and premalignant prostatic lesions. Am J Pathol 155: 641-647.
- Mobbs B, Johnson I, Liu Y (1990) Quantitation of cytosolic and nuclear estrogen and progesterone receptor in benign, untreated, and treated malignant human prostatic tissue by radioligand binding and enzyme?immunoassays. The Prostate 16: 235-244.
- Da J, Lu M, Wang Z (2015) Estrogen receptor alpha (ERα)-associated fibroblasts promote cell growth in prostate cancer. Cell Biochem Biophys 73: 793-798.
- ?lusarz A, Jackson GA, Day JK, Shenouda NS, Bogener JL, et al. (2012) Aggressive prostate cancer is prevented in ERαKO mice and stimulated in ERβKO TRAMP mice. Endocrinology 153: 4160-4170.
- Muthusamy S, Andersson S, Kim HJ, Butler R, Waage L, et al. (2011) Estrogen receptor β and 17β-hydroxysteroid dehydrogenase type 6, a growth regulatory pathway that is lost in prostate cancer. Proc Natl Acad Sci 108: 20090-20094.
- Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, et al. (2008) Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB j 22: 1512-1520.
- Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P (2004) Loss of ERβ expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer 11: 537-551.
- Hartman J, Ström A, Gustafsson JÅ (2012) Current concepts and significance of estrogen receptor β in prostate cancer. Steroids 77: 1262-1266.
- Lombardi AP, Pisolato R, Vicente CM, Lazari MF, Lucas TF, et al. (2016) Estrogen receptor beta (ERβ) mediates expression of β-catenin and proliferation in prostate cancer cell line PC-3. Mol Cell Endocrinol 430: 12-24.
- McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, et al. (2010) Estrogen receptor–β activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFα mediated. Proc Natl Acad Sci 107: 3123-3128.
- Siegel RL, Miller KD, Jemal A (2016) Cancer statistics.CA Cancer J Clin 66:7-30.
- Gjertson CK, Albertsen PC (2011) Use and assessment of PSA in prostate cancer. Med Clin North Am 95: 191-200.
- Attard G, Reid AH, Olmos D, de Bono JS (2009) Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res 69: 4937-4940.
- Shore ND, Abrahamsson PA, Anderson J, Crawford ED, Lange P (2013) New considerations for ADT in advanced prostate cancer and the emerging role of GnRH antagonists. Prostate Cancer Prostatic Dis 16:7-15.
- Sharifi N, Gulley JL, Dahut WL (2005) Androgen deprivation therapy for prostate cancer. JAMA 294: 238-244.
- Kelly K, Yin JJ (2008) Prostate cancer and metastasis initiating stem cells. Cell Res 18: 528-537.
- Nam RK, Sugar L, Yang W, Srivastava S, Klotz LH, et al. (2007) Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localised prostate cancer. Br J Cancer 97: 1690-1695.
- Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, et al. (2004) Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res 64: 9209-9216.
- Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, et al. (2000) Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol 31: 578-583.
- Craft N, Shostak Y, Carey M, Sawyers CL (1999) A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 5: 280-285.
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, et al. (2004) Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N Engl J Med 351: 1502-1512.
- Sharifi N, Gulley JL, Dahut WL (2010) An update on androgen deprivation therapy for prostate cancer. Endocr Relat Cancer 17: R305-315.
- Kirby M, Hirst C, Crawford ED (2011) Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract 65: 1180-1192.
- Afshar M, Evison F, James ND, Patel P (2015) Shifting paradigms in the estimation of survival for castration-resistant prostate cancer: A tertiary academic center experience. Urol Oncol 33: 338 e1-7.
- Ezzell EE, Chang KS, George BJ (2013) New agents in the arsenal to fight castrate-resistant prostate cancer. Curr Oncol Rep 15: 239-248.
- Schellhammer P, Sharifi R, Block N, Soloway M, Venner P, et al. (1996) A controlled trial of bicalutamide vs flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Casodex Combination Study Group. Urology 78: 745-752.
- Suzuki K, Nakazato H, Matsui H, Koike H, Okugi H, et al. (2003) Genetic polymorphisms of estrogen receptor alpha, CYP19, catechol-O-methyltransferase are associated with familial prostate carcinoma risk in a Japanese population. Cancer 98: 1411-1416.
- Ho SM (2004) Estrogens and anti-estrogens: key mediators of prostate carcinogenesis and new therapeutic candidates. J Cell Biochem 91: 491-503.
- Noble RL (1980) Production of Nb rat carcinoma of the dorsal prostate and response of estrogen-dependent transplants to sex hormones and tamoxifen. Cancer Res 40: 3547-3550.
- Bosland MC, Ford H, Horton L (1995) Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 beta or diethylstilbestrol. Carcinogenesis 16: 1311-1317.
- Ricke WA, McPherson SJ, Biancon JJ, Cunha GR, Wang Y, et al. (2008) Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J 22: 1512-1520.
- Bonkhoff H, Fixemer T, Hunsicker I, Remberger K (1999) Estrogen receptor expression in prostate cancer and premalignant prostatic lesions. Am J Pathol 155: 641-647.
- Lewis JS, Jordan VC (2005) Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat Res 591: 247-263.
- Meegan M, Lloyd D (2003) Advances in the science of estrogen receptor modulation. Curr Med Chem 10: 181-210.
- Bryant HU (2002) Selective estrogen receptor modulators. Rev Endocr Metab Disord 3: 231-241.
- Cano, Antonio, Calaf i Alsina, Joacquim, Duenas-Diez,et al (2006). Selective Estrogen Receptor Modulators A New Brand of Multitarget Drugs, ( New York: Springer).
- Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, et al. (1997) Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc Natl Acad Sci 94: 14105-14110.
- Macgregor JI, Jordan VC (1998) Basic guide to the mechanisms of antiestrogen action. Pharmacol Rev 50: 151-196.
- Jordan VC (2008) Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer 44: 30-38.
- Obiorah I, Jordan VC (2011) Progress in endocrine approaches to the treatment and prevention of breast cancer. Maturitas 70:315-321.
- Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F (2014) Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids 90:13-29.
- Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, et al. (2014) EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol 65:467-479.
- Serretta V, Altieri V, Morgia G, Nicolosi F, De Grande G, et al. (2012) A randomized trial comparing tamoxifen therapy vs. tamoxifen prophylaxis in bicalutamide-induced gynecomastia. Clin Genitourin Cancer 10:174-179.
- Rohrich RJ, Ha RY, Kenkel JM, Adams Jr WP (2003) Classification and management of gynecomastia: defining the role of ultrasound-assisted liposuction. Plast Reconstr Surg 111:909-923.
- Braunstein GD (1999) Aromatase and gynecomastia. Endocr Relat Cancer 6: 315-324.
- Davanço RA, Neto MS, Garcia ÉB, Matsuoka PK, Huijsmans JP, et al. (2009) Quality of life in the surgical treatment of gynecomastia. Aesthetic Plast Surg 33: 514-517.
- Barros ACSDd, Sampaio MdCM (2012) Gynecomastia: physiopathology, evaluation and treatment. Sao Paulo Med J 130:187-197.
- Wenger NK, Barrett-Connor E, Collins P, Grady D, Kornitzer M, et al. (2002) Baseline characteristics of participants in the Raloxifene Use for The Heart (RUTH) trial. Am J Cardio 90: 1204-1210.
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Wolmark N (2002) The study of tamoxifen and raloxifene: preliminary enrollment data from a randomized breast cancer risk reduction trial. Clin Breast Cancer 3: 153-159.
- Bryant HU, Dere WH (1998) Selective estrogen receptor modulators: an alternative to hormone replacement therapy. Exp Biol Med 217: 45-52.
- Morello KC, Wurz GT, DeGregorio MW (2003) Pharmacokinetics of selective estrogen receptor modulators. Clin Pharmacokinet 42: 361-372.
- Hochner-Celnikier D (1999) Pharmacokinetics of raloxifene and its clinical application. Eur J Obstet Gynecol Reprod Biol 85: 23-29.
- Kemp DC, Fan PW, Stevens JC (2002) Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab Disposition 30: 694-700.
- Jeong EJ, Lin H, Hu M (2004) Disposition mechanisms of raloxifene in the human intestinal Caco-2 model. J Pharmacol Exp Ther 310: 376-385.
- Stuart EC, Rosengren RJ (2008) The combination of raloxifene and epigallocatechin gallate suppresses growth and induces apoptosis in MDA-MB-231 cells. Life Sci 82: 943-948.
- Stuart EC, Jarvis RM, Rosengren RJ (2010) In vitro mechanism of action for the cytotoxicity elicited by the combination of epigallocatechin gallate and raloxifene in MDA-MB-231 cells. Oncol Rep 24: 779-785.
- Kim IY, Seong DH, Kim BC, Lee DK, Remaley AT, et al. (2002) Raloxifene, a selective estrogen receptor modulator, induces apoptosis in androgen-responsive human prostate cancer cell line LNCaP through an androgen-independent pathway. Cancer Res 62: 3649-3653.
- Kim HT, Kim BC, Kim IY, Mamura M, Seong DH, et al. (2002) Raloxifene, a mixed estrogen agonist/antagonist, induces apoptosis through cleavage of BAD in TSU-PR1 human cancer cells. J Biol Chem 277: 32510-32515.
- Shazer RL, Jain A, Galkin AV, Cinman N, Nguyen KN, et al. (2006) Raloxifene, an oestrogen?receptor?beta?targeted therapy, inhibits androgen?independent prostate cancer growth: results from preclinical studies and a pilot phase II clinical trial. BJU international 97: 691-697.
- Olivier S, Close P, Castermans E, De Leval L, Tabruyn S, et al. (2006) Raloxifene-induced myeloma cell apoptosis: a study of nuclear factor-kappaB inhibition and gene expression signature. Mol Pharmacol 69: 1615-1623.
- Ball LJ, Levy N, Zhao X, Griffin C, Tagliaferri M, et al. (2009) Cell type- and estrogen receptor-subtype specific regulation of selective estrogen receptor modulator regulatory elements. Mol Cell Endocrinol 299: 204-211.
- Kim IY, Seong DH, Kim BC, Lee DK, Remaley AT, et al. (2002) Raloxifene, a selective estrogen receptor modulator, induces apoptosis in androgen-responsive human prostate cancer cell line LNCaP through an androgen-independent pathway. Cancer Res 62: 3649-3653.
- Piccolella M, Crippa V, Messi E, Tetel MJ, Poletti A (2014) Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacol Res 79: 13-20.
- Rossi V, Bellastella G, De Rosa C, Abbondanza C, Visconti D, et al. (2011) Raloxifene induces cell death and inhibits proliferation through multiple signaling pathways in prostate cancer cells expressing different levels of estrogen receptor alpha and beta. J Cell Physiol 226: 1334-1339.
- Neubauer BL, Best KL, Counts DF, Goode RL, Hoover DM, et al. (1995) Raloxifene (LY156758) produces antimetastatic responses and extends survival in the PAIII rat prostatic adenocarcinoma model. Prostate 27: 220-229.
- Palmer, Hannah (2015) Using raloxifene and the curcumin derivative, RL91, for the treatment of castrate resistant prostate cancer. MSc Thesis, University of Otago.
- Zeng Y, Yokohira M, Saoo K, Takeuchi H, Chen Y, et al. (2005) Inhibition of prostate carcinogenesis in probasin/SV40 T antigen transgenic rats by raloxifene, an antiestrogen with anti-androgen action, but not nimesulide, a selective cyclooxygenase-2 inhibitor. Carcinogenesis 26: 1109-1116.
- Shazer RL, Jain, A, Galkin, AV, Cinman, N, Nguyen, KN, et al. (2006) Raloxifene, an oestrogen-receptor-beta-targeted therapy, inhibits androgen-independent prostate cancer growth: results from preclinical studies and a pilot phase II clinical trial. BJU Int 97: 691-697.
- Smith MR, Fallon MA, Lee H, Finkelstein JS (2004) Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial. J Clin Endocrinol Metab 89: 3841-3846.
- Ho TH, Nunez-Nateras R, Hou YX, Bryce AH, Northfelt DW, et al. (2016) A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clin Genitourin Cancer 15: 196-202.
- Shoda T, Kato M, Fujisato T, Misawa T, Demizu Y, et al. (2016) Synthesis and evaluation of raloxifene derivatives as a selective estrogen receptor down-regulator. Bioorg Med Chem 24: 2914-2919.
- Ning M, Zhou C, Weng J, Zhang S, Chen D, et al. (2007) Biological activities of a novel selective oestrogen receptor modulator derived from raloxifene (Y134). Br J Pharmacol 150: 19-28.
- Vinceti M, Dennert G, Crespi CM, Zwahlen M, Brinkman M, et al. (2014) Selenium for preventing cancer. Cochrane Database Syst Rev 30: CD005195.
- Arsenyan P, Paegle E, Domracheva I, Gulbe A, Kanepe-Lapsa I, et al. (2014) Selenium analogues of raloxifene as promising antiproliferative agents in treatment of breast cancer. Eur J Med Chem 87: 471-483.
- Dadiboyena S (2012) Recent advances in the synthesis of raloxifene: a selective estrogen receptor modulator. Eur J Med Chem 51: 17-34.
- Aditya N, Ravi PR, Avula US, Vats R (2014) Poly (epsilon-caprolactone) nanocapsules for oral delivery of raloxifene: process optimization by hybrid design approach, in vitro and in vivo evaluation. J Microencapsul 31: 508-518.
- Fontana MC, Beckenkamp A, Buffon A, Beck RC (2014) Controlled release of raloxifene by nanoencapsulation: effect on in vitro antiproliferative activity of human breast cancer cells. Int J Nanomedicine 9: 2979-2991.
- Kushwaha AK, Vuddanda PR, Karunanidhi P, Singh SK, Singh S (2013) Development and Evaluation of Solid Lipid Nanoparticles of Raloxifene Hydrochloride for Enhanced Bioavailability. Biomed Res Int 2013:584549.
- Ravi PR, Aditya N, Kathuria H, Malekar S,Vats R (2014) Lipid nanoparticles for oral delivery of raloxifene: optimization, stability, in vivo evaluation and uptake mechanism. Eur J Pharm Biopharm 87: 114-124.
- Saini D, Fazil M, Ali MM, Baboota S, Ali J (2015) Formulation, development and optimization of raloxifene-loaded chitosan nanoparticles for treatment of osteoporosis. Drug Deliv 22: 823-836.
- Tran TH, Poudel BK, Marasini N, Chi SC, Choi HG, et al. (2013) Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Int J Pharm 443: 50-57.
- Taurin S, Nehoff H, van Aswegen T, Rosengren RJ, Greish K (2014) A Novel Role for Raloxifene Nanomicelles in Management of Castrate Resistant Prostate Cancer. Biomed Res Int 2014:323-594.
- Pritchard T, Rosengren RJ, Greish K, Taurin S (2016) Raloxifene nanomicelles reduce the growth of castrate-resistant prostate cancer. J Drug Target 24: 441-449.