Research Article
Snaefridur Halldorsdottir1,2,#, Una Bjarnadottir1,#, Laufey Geirsdottir1,2, Brynja Gunnlaugsdottir1,2 and Bjorn Runar Ludviksson1,2,*
1Department of Immunology, Landspitali - The National University Hospital of Iceland, Reykjavík, Iceland
2Faculty of Medicine, Biomedical Center, University of Iceland, Reykjavík, Iceland
#These authors contributed equally to this work
Corresponding author
Dr. Bjorn Runar Ludviksson, Professor, Department of Immunology, Landspitali - The National University Hospital of Iceland, Building #14 at Eiriksgata, 101 Reykjavik, Iceland, Tel: 354 543 5810; Fax 354 525 4886; E-mail: bjornlud@landspitali.is
Received Date: 23rdJuly 2014
Accepted Date: 27th August 2014
Published Date: 28th August 2014
Citation
Ludviksson BR (2014) TNF? and IL-1? have dose dependent effects on differentiation of CD4+CD127-CD25highFoxP3high T cells. Enliven: Immunol Immunotechnol 1(1):001
Copyright
@ 2014 Dr. Bjorn Runar Ludviksson. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, that permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Regulatory T cells (TRegs) are critical for the maintenance of balanced human immune responses. The innate immune system plays a significant role in the pathogenesis of autoimmune diseases. However, its role in the differentiation and function of human CD4+ induced TRegs (iTRegs) is currently unclear. In this study we investigated the effect of the pro-inflammatory cytokines tumor necrosis factor alpha (TNFα) and IL-1β upon the differentiation and function of human CD4+FoxP3+ iTRegs.
Naïve human CD4+CD25- T cells, isolated from both cord blood (CB) and adult peripheral blood (PB) were cultured under various stimulatory conditions with/without TNFα and IL-1β. The CD4+FoxP3+ iTRegs were characterized as CD4+CD127-CD25highFoxP3high iTRegs and their suppressive function evaluated.
TNFα and IL-1β significantly affected the differentiation of human CD4+FoxP3+ iTRegs in a dose and time dependent manner. Low dose and short term conditions promoted their differentiation while high dose and long term conditions were inhibitory. In addition, the suppressive function of the CD4+FoxP3+ iTRegs was inhibited by TNFα and IL-1β. This inhibitory effect of TNFα and IL-1β was associated with a significant reduction of the transforming growth factor β receptor type II (TβRII) expression and IL-2 secretion. However, their IL-10 secretion was not affected and no IL-35 secretion was observed in any of the culture conditions tested.
We conclude that the in vitro induction of human CD4+FoxP3+ iTRegs is mediated through TβRII and IL-2 dependent mechanism. The regulatory effect of TNFα and IL-1β on CD4+FoxP3+ iTReg differentiation is dynamic and may to some extent reflect the magnitude and persistence of the initial pro-inflammatory response.
Abbreviations
CB: Cord Blood; FoxP3: Forkhead box P3; iTRegs: Induced regulatory T cells; nTRegs: Natural Regulatory T cells; PB: Peripheral Blood; RA: Rheumatoid Arthritis; TGF-?1: Transforming Growth Factor ?1; T?RII: Transforming Growth Factor ? Receptor Type II; TNF?: Tumor Necrosis Factor ?; TNFRI: Tumor Necrosis Factor Receptor Type I; TNFRII: Tumor Necrosis Factor Receptor Type II; TRegs: Regulatory T cells
Introduction
TRegs are vital for immune suppression and crucial for controlling inflammatory responses, establishing self-tolerance and maintaining immune homeostasis[1].CD4+ TRegs are divided into two major classes; naturally occurring TRegs (nTRegs) and iTRegs that are phenotypically indistinguishable but may differ in functionality and differentiation [2,3].
Inducible TRegs develop in the periphery from CD4+CD25- T cells in the presence of transforming growth factor (TGF)-?1 [4,5] and IL-2 [6]. They are essential for maintaining an effective peripheral immune tolerance and are characterized by high surface expression of the IL-2 ? receptor chain (CD25), low expression of IL-7 receptor ? (CD127) and stable expression of the transcription factor, forkhead-box protein P3 (FoxP3) [7, 8]. FoxP3 has been used as the most specific TRegs marker and is required for normal TReg function and differentiation [9]. Therefore, prolonged FoxP3 expression is known to be required to define TRegs indisputably [10].It is well documented that TGF-?1 is essential for T cell homeostasis. This has been established in TGF-?1 knockout mice, which develop various inflammatory autoimmune diseases [11]. Furthermore, TGF-? [12,13], prostaglandin E2 [14] and retinoic acid [15,16] are the only recognized inducers of FoxP3. Therefore, TGF-?1 has a great therapeutic potential, both as the generator of TRegs [17] and possibly also as the main mediator of their immunosuppressive activity [18]. Our results have shown that T cells are more responsive towards TGF-?1 mediated suppression during suboptimal stimulatory conditions [19]. Accordingly, autoreactive T cells that drive chronic inflammatory responses in autoimmune diseases are probably highly stimulated and therefore unresponsive towards the immune-modifying effects of TGF-?1.
TNF? is a pro-inflammatory cytokine with a capacity to induce apoptosis. Furthermore, it plays an important role in different biological processes including the induction of other cytokines. TNF? is a key factor in numerous inflammatory diseases.The success of anti-TNF biologicals for the treatment of rheumatoid arthritis (RA) [20], Crohn´s disease and other chronic inflammatory conditions (reviewed in [21]) highlights TNF? as a key mediator of inflammation [22]. TRegs expressing membrane bound-TNF? have been linked to severe RA disease activity [23] and it has been reported that neutralization of TNF? restored TReg function and numbers [24].Paradoxically, some researchers have also shown that TNF? regulates the function and numbers of TRegs [25].Recent study has additionally shown that TRegs in mice and humans are able to shed massive amounts of the TNF receptor type II (TNFRII) after few days of activation [22].This can possibly explain the contradictory findings regarding the effect of TNF? on TReg function and numbers as described above. However, most studies agree that TNF? plays an important role in TReg activity, although the exact mechanism is currently unclear.
IL-1? is a pro-inflammatory cytokine, synthesized as a precursor molecule (pro-IL-1?) by many different cell types (reviewed in ref. [26]). It is involved in the pathogenesis of inflammatory diseases and a number of autoimmune diseases are associated with high levels of IL-1? [27,28]. Additionally, it is an important contributor to the polarization of Th17 cells [29,30]. Despite its possible role in TReg differentiation in mice [31], the effect of IL-1? on human TRegs remains to be explored. Given the protective role of the pro-inflammatory cytokines TNF? and IL-1? during infection, and the fact that both cytokines have been reported to antagonize TGF-?1, we hypothesized that these cytokines affect the IL-2 and TGF-?1 mediated induction of iTRegs. Therefore, the aim of the study was to evaluate how stimulatory conditions and pro-inflammatory cytokines influence the ability of naïve human CD4+CD25- T cells to differentiate into functional CD4+ iTRegs (CD4+CD25highCD127-FoxP3high) from naïve T cells.
Materials and Methods
Study Material
The study was approved by The Ethics Committee of Landspitali, University Hospital of Iceland and The Data Protection Authority of Iceland. All human samples were obtained after a written informed consent had been obtained from the donors. CB was obtained after normal deliveries at the Department of Obstetrics & Genecology at Landspitali, University Hospital of Iceland. Mothers with autoimmune disorders were excluded from the study. Adult venous PB was collected from healthy volunteers at the Icelandic Blood Bank.
Isolation of CD4+CD25- T cells
Mononuclear cells (PB and CB) were isolated from heparinized blood by density gradient centrifugation over Ficoll-Hypaque (Sigma-Aldrich) at room temperature for 30 min. The cells were centrifuged at 1600 rpm. CD4+ T cells were isolated with Dynabeads CD4 (Dynal, Invitrogen) according to the manufactures instructions. CD25+ T cells were depleted from the CD4+ T cell population using Dynabeads CD25 (Dynal, Invitrogen). The purity of the isolated CD4+CD25- T cells was consistently >95% and their phenotypes, determined by flow cytometry after staining for the surface markers, are listed in (Table 1).
| CD103 | CD25 | CD28 | FoxP3 | T?RII | CD127 | CD45RA | CD45RO | CD4+CD25highCD127-FoxP3high | |
| N | ?3 | ?3 | ?3 | ?3 | ?3 | ?3 | ?3 | ?3 | ?3 |
| Mean (%) | 4.22 | 0.05 | 85.88 | 1.89 | 12.1 | 0.06 | 81.6 | 8.6 | 1.08 |
| SEM | 2.24 | 0.02 | 6.25 | 0.71 | 2.07 | 0.03 | 2.34 | 7 | 0.13 |
Table 1: Phenotypic characterization of CD4+CD25- T cells after isolation.
Induction of TRegs
T cells (CD4+CD25-) were stimulated with plate-bound anti-CD3? (100 µL, 1 µg/mL) monoclonal antibody (UCHT) and soluble anti-CD28 (1 µg/mL, 37407), unless otherwise stated, for 24-120 hrs in the presence of IL-2 (100 IU) and with or without TGF-?1 (10 ng/mL). Various concentration of TNF? (0.5-50 ng/mL) and IL-1? (0.1-10 ?g/mL) were added into selected cultures (antibodies and cytokines from R&D Systems Inc). The CD4+CD25- T cells (1x106 /mL) were cultured in a serum free medium (AimV, Nunc, Invitrogen) in 96 well U-bottomed cell culture plates (Nunc, Invitrogen) and incubated at 37°C under a 5% CO2 and 95% humidity in a cell culture system incubator.
Surface Markers and Intracellular Staining
The cells were washed with staining buffer (PBS with 2 mM EDTA, 0.5% BSA and 0.1% sodium aside) and incubated for 20 min at 4°C with fluorescent antibodies. After staining, the samples were washed and re-suspended in staining buffer. The cells were either fixed in PBS containing 0.5% formalin and stored for up to a week at 4°C or subjected immediately to FACS. A total of 100,000 events were collected in the lymphocyte gate and the data analyzed using either CellQuest (BD Biosciences) or FlowJo (Tree Star Software). The cells were fixed and permeabilized with FoxP3 staining set (eBiosciences) and stained with a FoxP3 specific antibody (clone 236A/E7, eBiosciences) or matched isotype control according to the manufacturer?s instructions. The cells were analyzed immediately after staining.
The following FITC, PE, PerCP, PerCP-Cy5.5 and APC conjugated human antibodies were used for flow cytometric analysis; CD4 (RPA-T4), CD25 (BC96), CD103 (Ber-ACT8), CD45RA (HI100), CD45RO (UCHL1), T?RII (polyclonal goat anti-human), TNFRI/II, (16803, 25508), CD127 (eBioRDR5) and FoxP3 (236A/E7). Appropriate isotype controls were used to set the quadrants and to evaluate background staining. The antibodies were purchased from BD Biosciences, R&D Systems, eBiosciences and Biolegend.
Proliferation Assay
To assess T cell proliferation, CD4+ T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) prior to stimulation. After washing the cells twice in 10 mL PBS, they were incubated with 0.5 mM CFSE in 1 mL PBS per 1x107 cells at room temperature for 8 min. To stop the staining reaction, 8 mL fetal calf serum (FCS) was added. The cells were then washed twice in 10 mL AimV medium containing 5% FCS and re-suspended in the appropriate volume of medium (1x106 /mL). The CD4+ T cell proliferation was assessed by flow cytometric analysis of the CFSE dilution.
Suppression Assay
The CD4+CD25- T cells were stimulated under iTReg inducing conditions for 120 hrs and their suppressive function evaluated. The CD4+ iTRegs were harvested and co-cultured for 72 hrs with CFSE labeled allogeneic PBMCs and Epstein-Barr transformed B cells (EB-B cells) in AimV at various ratios to assess T cell proliferation [32,33]. The EB-B cells (2x106 /mL) had previously been exposed to superantigens (Staphylococcal enterotoxins, SEA, SEB and SEE, Toxin Technologies,1 µg/mL of each) for 2 hrs and washed 3 times in PBS. The ratio between PBMCs:EB-B cells and between iTRegs and EB-B cells was constant at 10:1 whereas the ratio of iTRegs: PBMCs varied from 1:1 to 1:32. The T cell proliferation of the CFSE labeled cells were assessed by BD FACSCalibur. Cells were gated on CD25+ lymphocytes and the percentages of cells in each generation were calculated by Modfit LT software. The proliferation index (PI) was determined as the sum of the cells in all generations divided by the computed number of original parent cells theoretically present at the start of the experiment.
In Statistical analysis, it is better to include more information in ?allergenic PBMC and EB-B cell? and why these PBMC proliferation?
ELISA
After incubation the cell cultures were centrifuged and cell culture supernatants collected and stored at -80°C. Total TNF?, sTNFRII and IL-10 were measured in the supernatants using DuoSet ELISA kits (DY210 and DY726, R&D Systems). IL-2 and IL-35 were measured using Human IL-2 ELISA Set (BD Biosciences) and ELISA Ready-Set-Go! (eBioscience).
Statistical analysis
Statistical analyses were performed using GraphPad Prism (Version 5.04 GraphPad Sofware Inc, for windows, La Jolla, CA, USA, www.graphpad.com ). Paired student?s t-test was used and results expressed as mean values ± standard error of the mean (SEM). Differences were determined to be significant when P < 0.05 (two-tailed).
Results
FoxP3 Expression is Increased in Response to T Cell Activation
It is well documented that higher amount of naïve T cells are found in CB than adult PB due to limited exposure of CB T cells to exogenous antigens [32-34]. Furthermore, this naïve phenotype has been implied to possess a significantly enhanced proliferation [35]. Therefore, the induction of FoxP3 expression was initially evaluated in CD4+CD25- T cells isolated from healthy human umbilical CB. Naïve CD4+CD25- cells were isolated and stimulated for 72 hrs and iTRegs were identified as CD4+CD127-CD25highFoxP3high using the gating strategy displayed in (Figure 1).
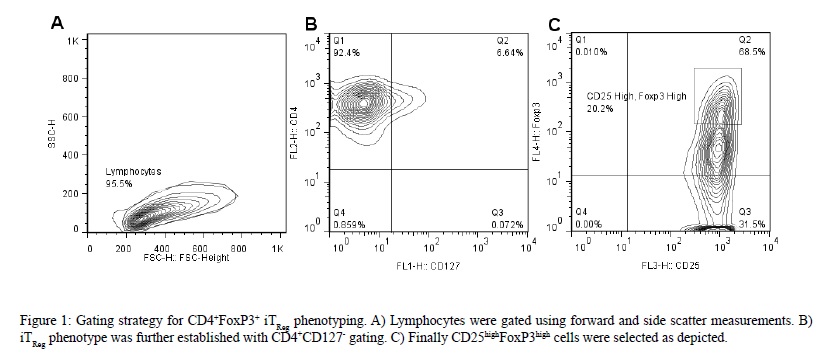
As shown in (Figure 2A), the maximum CD4+CD127-CD25highFoxP3high iTRegs in vitro inducing conditions were obtained when the cells were stimulated with high dose anti-CD3 (10 µg/mL) and anti-CD28 in the presence of TGF-?1 and IL-2 for 72 hrs (84%±4.3). In contrast, low intensity T cell receptor (TCR) stimulation (anti-CD3, 1 µg/mL) with CD28 co-stimulation was not as strong inducer of CD4+CD127-CD25highFoxP3high iTReg differentiation (29.5% reduction, P < 0.001; Figure 2A). Therefore, during our next set of experiments the low dose stimulation model was used to further evaluate possible additional effect of pro-inflammatory cytokines on the differentiation of CD4+CD127-CD25highFoxP3high iTRegs.
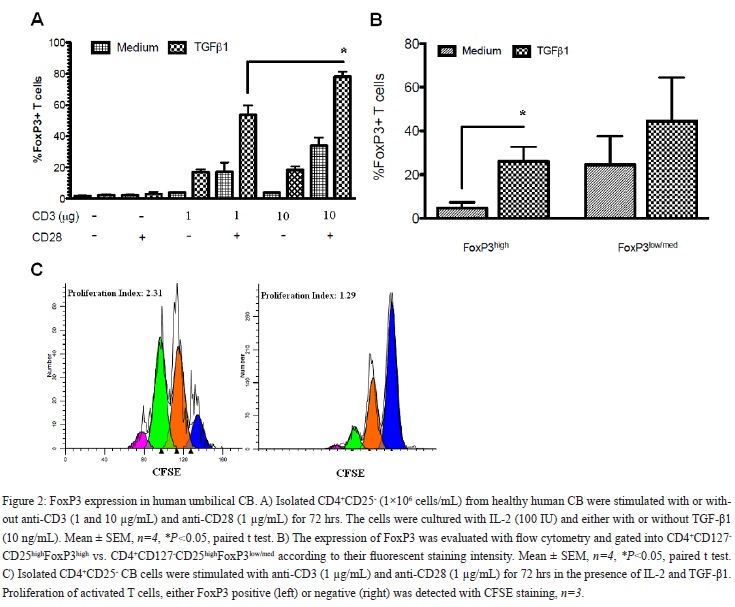
Comparison of CD4+CD127-CD25highFoxP3low/med (nonTRegs), which are not functionally active and do not suppress effector cells, vs. the CD4+CD127-CD25highFoxP3high iTRegs showed that only the latter population expanded in the presence of TGF-?1 (% iTRegs: 4.6%±2.7 iTRegs without TGF-?1 vs. 26%±6.8 with TGF-?1; P < 0.001; Figure 2B). A major consequence of active TGF-?1 signaling is inhibition of CD4+ T cell proliferation [36]. Therefore, we tested whether TGF-?1 had the same effect on FoxP3+ cells during their primary cellular response and differentiation. As shown in (Figure 2C), proliferation of FoxP3negative CD4+ T cells was inhibited in the presence of TGF-?1 with less than 15% of the cells going through two or more divisions. However, proliferation of induced FoxP3+ positive cells was not decreased in the presence of TGF-β1 with an average of 68% of the cells dividing at least twice [Figure 2C]. Therefore, the main focus was on the CD4+CD127-CD25highFoxP3high population in our remaining studies and they will be defined as CD4+FoxP3+ iTRegs cells from now on for simplicity.
In [Figure 2B], what kind of cells does Foxp3low/med refers to? In [Figure 2C], how about the proliferation of Foxp3low/med+ cells? This should be discussed.
The Dual Effect of TNF? on FoxP3 Expression
TGF-?1 has preferentially a positive effect on the differentiation of iTRegs and TNFRII expression has been associated with nTRegs. Therefore, we examined the effect of TNF? upon CD4+FoxP3+ iTReg differentiation. To evaluate if the magnitude of pro-inflammatory responses would influence CD4+FoxP3+ iTReg differentiation, CD4+CD25- T cells, from both PB and CB (data not shown), were cultured with low and high dose TNF?. As shown in [Figure 3], prolonged exposure of high dose TNF? had a negative effect on CD4+FoxP3+ iTReg differentiation. In contrast, low dose TNF? (0.5/5.0 ng/mL) had no effect upon CD4+FoxP3+ iTReg differentiation (data not shown). Therefore, the effects of TNF? on CD4+FoxP3+ iTReg differentiation are dose dependent for T cells isolated from both PB and CB.
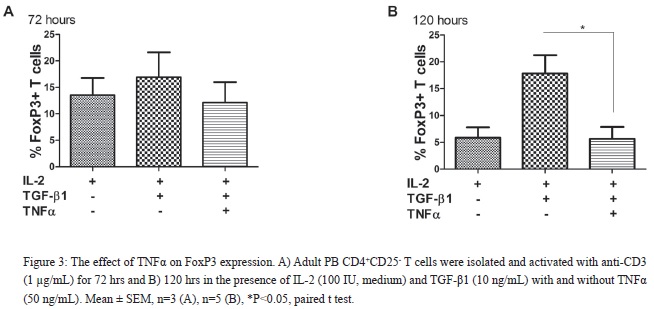
In (Figure3), the authors used very high dose of TNF-a (50 ng/ml)? What is the physiological concentration of TNF-a? What is the effect of TNFa on 5-10 ng/ml?
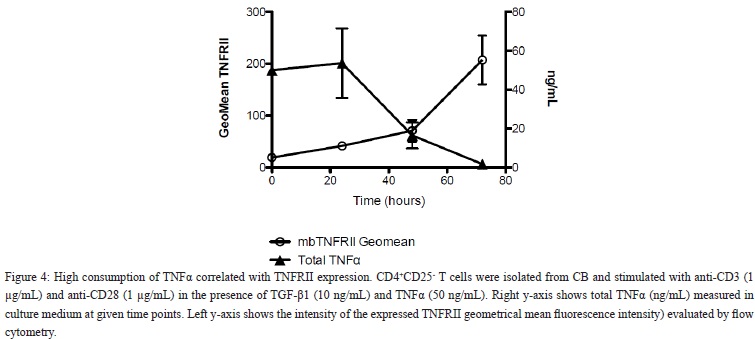
High Consumption of TNF? Correlated With Up-Regulation of TNFRII Bound Receptor
Naïve CB CD4+CD25- T cells do not express TNFRII. However, when stimulated with anti-CD3, anti-CD28 and IL-2, TNFRII receptor expression is up-regulated after 48 hrs of culture (data not shown). In order to estimate the consumption of TNF? by the T cells, the TNF? in the culture supernatant was measured. High consumption of TNF? was detected in the cultures that reached its maximum effect at 72 hrs. Thus, high dose of exogenously added TNF? (50 ng/mL) had mostly been consumed from the culture medium within 72 hrs [Figure 4]. The rate of disappearance of TNF? in culture medium correlated negatively with high TNFRII expression per cell (r=-0.759; P=0.002; Figure 4). Furthermore, during in vitro induction of CD4+FoxP3+ iTRegs from CD4+CD25- T cells TNF? was negatively correlated with the expression of cell bound TNFRII.
The Effect of IL-1? on FoxP3 Expression
The effect of IL-1? on the differentiation of CB CD4+CD25- T cells into CD4+FoxP3+ iTRegs was evaluated as well. Short term exposure to low dose of IL-1? (0.1 ng/mL) induced a two-fold increase of differentiated CD4+FoxP3+ iTRegs compared to TGF-?1 alone (Figure 5A, P < 0.05). In contrast, long term (120 hrs) and high dose of IL-1? (10 ng/mL), significantly decreased the differentiation of CD4+FoxP3+ iTRegs (73.5% reduction, P < 0.05; Figure 5B). Adult PB CD4+CD25- T cells responded similarly to prolonged exposure to high dose of IL-1?. As shown in Figure 5C-5D, IL-1? significantly reduced the proportion of CD4+FoxP3+ iTRegs induced from adult T cells after 72 and 120 hr exposure. In addition, low dose of IL-1? had no significant effects upon CD4+FoxP3+ iTReg differentiation in adult PB (data not shown). These findings suggest that the effects of the pro-inflammatory cytokine IL-1? on CD4+FoxP3+ iTReg differentiation depend upon the cytokine dose and time of exposure.
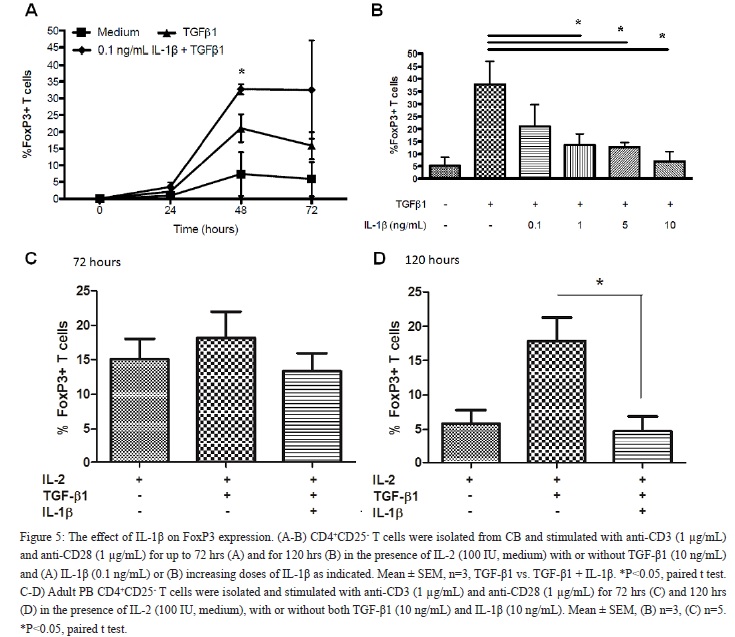
IL-1? Inhibits T?RII Expression
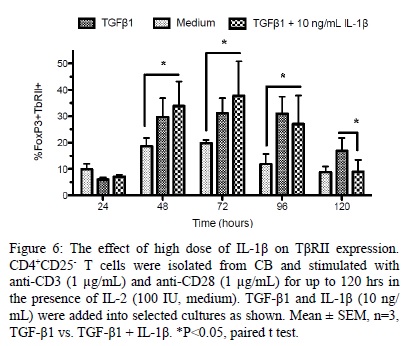
We next evaluated the effect of IL-1? upon T?RII expression. As shown in [Figure 6], high dose of IL-1? significantly inhibited the expression of T?RII on CD4+FoxP3+ iTRegs following long term (120 hrs) stimulation (46 ± 3.6% reduction, P < 0.05) compared with cells cultured with TGF-?1 alone.
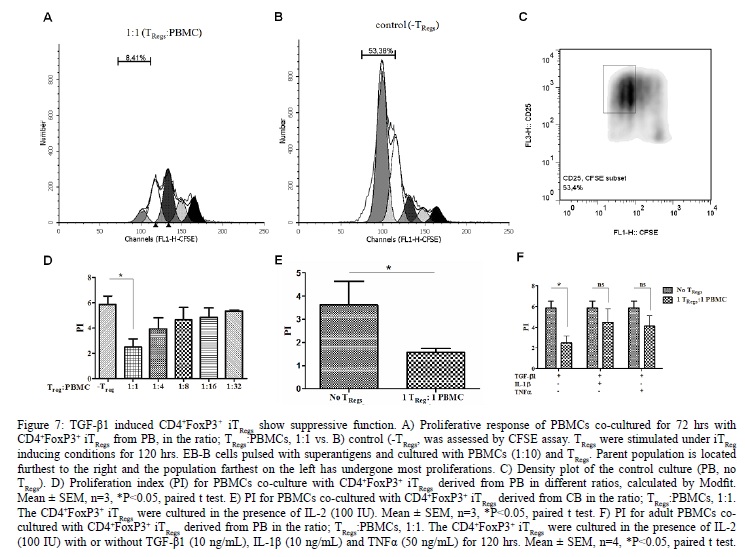
IL-1? and TNF? Inhibited the Suppressive Function of iTRegs
The functionality of the CD4+FoxP3+ iTRegs was evaluated using a previously established superantigen driven culture system [3]. As shown in [Figure 7] the CD4+FoxP3+ iTRegs from PB had a significant suppressive effect. In the presence of CD4+FoxP3+ iTRegs [Figure 7A], very little proliferation (8.4%) was observed however in the absence of CD4+FoxP3+ iTRegs [Figure 7B/C] much higher proportion (53.4%) of the CFSE stained PBMCs proliferated. Six-fold reduction [Figure 7D] in PBMCs proliferation in the presence of CD4+FoxP3+ iTRegs (1:1 ratio, P < 0.05) was observed. This was also true for CD4+FoxP3+ iTRegs derived from CB [Figure 7E]. The suppressive function of in vitro CD4+FoxP3+ iTRegs correlated positively with their numbers [Figure 7D]. Furthermore, both IL-1? and TNF? were found to reduce suppressive function after long term and high dose induction [Figure 7F].
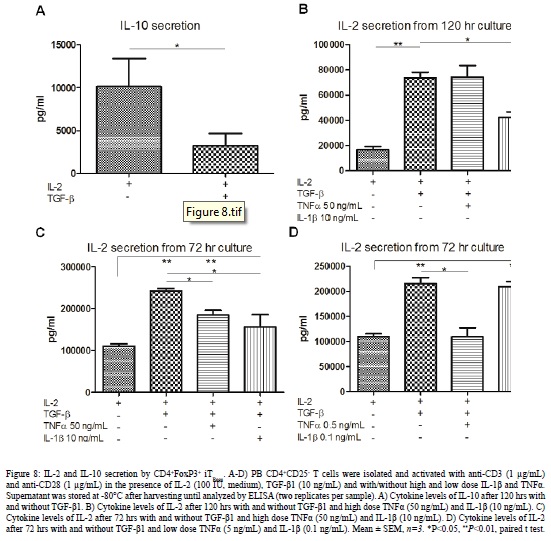
Cytokine Secretion by iTRegs
The cytokine secretion of PB CD4+FoxP3+ iTRegs was evaluated after 72 and 120 hr culture. As shown in [Figure 8], differentiated CD4+FoxP3+ iTRegs cells secreted significantly lower amounts of IL-10 compared to controls after 120 hrs (Figure 8A). The pro-inflammatory cytokines had no significant effects on the IL-10 secretion (data not shown). However, the CD4+FoxP3+ iTRegs secreted significantly more IL-2 in the presence of TGF-?1. Thus, we investigated the effects of the pro-inflammatory cytokines on the IL-2 secretion. High dose IL-1? had significant effects on IL-2 secretion compared to TGF-? alone (Figure 8B-8C). High dose TNF? showed similar effects after 72 hrs culture (Figure 8C) but the effects were lost after 120 hrs (Figure 8B). Same results were obtained with CD4+FoxP3+ iTRegs cultured from CB (data not shown). On the other hand, low dose IL-1? did not affect the IL-2 secretion of CD4+FoxP3+ iTRegs whereas low dose TNF? reduced IL-2 secretion (Figure 8D). Negligible amounts of IL-35 were detected at all culture conditions tested (data not shown). Hence, these data suggest that CD4+FoxP3+ iTRegs mediate their suppressive function through IL-2 secretion and that IL-1? mediates its negative effects via IL-2 inhibition.
Discussion
In this study we have shown that the pro-inflammatory cytokines TNF? and IL-1? regulate the IL-2 and TGF-?1 mediated differentiation of human CD4+FoxP3+iTRegs. The regulatory effects were found to be time and dose dependent. We also demonstrate that adult human CD4+CD25- T cells stimulated in the presence of IL-2 and TGF-?1 acquired a regulatory function that was lost if high doses of either TNF? or IL-1? were present during the induction phase. Finally, our results suggest that this could be driven through T?RII expression via IL-2 dependent, but IL-10 and IL-35 independent mechanism. However, further studies are needed in order to prove such a mechanism.
TNF? has been recognized as a key inducer of pathological immune responses in RA and several other autoimmune disorders. The efficacy of anti-TNF? treatment in RA has been associated with restored TReg numbers and function [34,35]. In addition, we have previously demonstrated that the anti-inflammatory effect of anti-TNF? antibody (Infliximab) is in part mediated through TGF-?1 dependent mechanism that is antagonized through TNF? [36]. However, a significant proportion of RA patients do not respond to this treatment and the cases of adverse reaction to this treatment indicate that TNF? is not consistently associated with a reduced numbers or function of TRegs [37]. Moreover, the induction of a lupus like syndrome in a few cases suggests that TNF? may promote self tolerance in selected individuals [37]. These findings are supported by recent human and murine studies showing that TNF? can either positively or negatively affect TReg induction and function. Kleijwegt et al. demonstrated positive effects of TNF? on the induction of human TRegs in vitro [38]. They observed that a membrane bound TNF? expressed by dendritic cells was critical for the in vitro induction of human TRegs. In contrast, it has been reported that TNF? negatively affects the induction of human TRegs in vitro [34,39]. Similarly, controversial results regarding the effect of TNF? on TRegs have been obtained. In that regard, it has been demonstrated that TNF? negatively affected FoxP3 expression and suppressive function of human TRegs in vitro [24]. In contrast, several studies suggest that TNF? promotes the function of TRegs. For instance, Chen et al. reported that TNF? expanded and enhanced the suppressive function of murine TRegs [25], a process dependent on TNFRII. Furthermore, another study demonstrated that TNF? secreted by pathogenic T effector cells enhanced the tolerogenic potential of TRegs that were co-transferred into mice, suffering from autoimmune diabetes [40].
In light of these findings our results are of particular interest as they underline the importance of the exogenous TNF? dosage. Thus, in our study, a low dose of TNF? (0.5 ng/mL) promoted the induction CD4+FoxP3+ iTRegs, whereas, a high dose (50 ng/mL) reduced their induction. Furthermore, we observed that this positive effect was time-dependent and only present early in the induction phase (Figure 4).
Negative effect of high dose TNF? (50 ng/mL) on the differentiation of CD4+ iTRegs has been reported by others [24,39]. In addition, studies using lower dose of TNF? (5-20 ng/mL) did not observe any negative effects upon iTReg differentiation [41,42]. Moreover, it has been demonstrated that high dose of TNF? has antagonistic effect upon the anti-inflammatory role of TGF-?1 on human CD4+ T cells [36].
The role of IL-1? signaling during iTReg differentiation is not clear. IL-1? is known to increase proliferation of conventional T cells and studies in mice have also shown that IL-1? enhances expansion of FoxP3+ T cells [31]. Conversely, IL-1? has been shown to negatively regulate or switch the phenotype of TRegs [30,43]. In support of this finding, IRAK-/- mice (lacking functional IL-1 signaling) have been reported to have higher TReg proportions compared with wild type mice [44]. This finding suggests that IL-1 signaling negatively regulates TReg development or maintenance. Similarly, IL-1?, in combination with IL-2 was recently shown to convert human nTRegs into Th17 lineage cells [30]. The effect of IL-1? on differentiating human iTRegs, has however to our knowledge, not been reported previously [45].
Our findings suggest that exogenously added IL-1? can either enhance or reduce the induction of CD4+FoxP3+ iTRegs, depending on the dose applied. Thus, a low dose of IL-1? (0.1 ng/mL) induced significant differentiation of CD4+FoxP3+ iTRegs, whereas, a high dose of IL-1? (5-10 ng/mL) had a reverse effect. Therefore, our results suggest that both TNF? and IL-1? have a time and dose dependent effect on the IL-2 and TGF-?1 mediated induction of human CD4+FoxP3+ iTRegs. Interestingly, the suppressive effects of IL-1? on FoxP3 induction were associated with a significant reduction in T?RII expression. Therefore, these results suggest that the negative effects of IL-1? upon FoxP3 expression may partially be caused by the reduced expression of TβRII and consequently reduced TGF-? signaling. These findings indicate that innate immunity may play a major role in defining its final outcome into either a tolerogenic or prolonged inflammatory response.
TGF-β1 in synergy with IL-2 is known to be required for effective FoxP3 expression. FoxP3 is the most consistent marker for human as well as murine TRegs. However, the expression of FoxP3 by human T cells is not consistently associated with a suppressive function [10]. TGF-β1 is also known to have anti-proliferative effects upon T cells. Our results from experiments studying the effect of TGF-β1 and IL-2 on proliferation of CD4+CD25+ T cells showed that FoxP3high cells are not affected by the anti-proliferative effect of TGF-β1. To confirm that this was not only due to stimulation and the presence of IL-2, we showed that FoxP3low/med cells do not proliferate in the presence of TGF-β1 in the same stimulatory conditions. This could possibly be due to innate bias of the T cells to differentiate into iTRegs in this stimulatory environment, or at least in the presence of low concentrations of antigens. Furthermore, human CD4+CD25- T cells stimulated in the presence of IL-2 and TGF-?1 have either been shown to be suppressive [46] or not, despite expressing high levels of FoxP3 [47]. However, our findings clearly demonstrate that human CD4+FoxP3+ iTRegs can be suppressive but this function was lost if high dose TNF? or IL-1? was present during their induction phase.
It has been suggested that TRegs compete with their responder cells for IL-2 consumption in mice and thus inhibit their proliferation [48]. However, IL-2 has been shown to be essential for TReg efficient suppressor function in vitro [49]. In our model we detected enhanced IL-2 secretion in the presence of TGF-?1. The high IL-2 cannot be explained by its administration as its half-life is 1-2 h and therefore our administrative amount of IL-2 has been diminished after 120 h in culture [50]. We also observed that after 120 hrs of culture, the presence of IL-1? had a significant negative effect on IL-2 secretion. In regard to our previous results of the negative effects of IL-1? on the CD4+FoxP3+ iTReg differentiation and function, we speculate that IL-1? mediates its negative effects through IL-2 inhibition. Although low and high dose TNF? significantly decreased the IL-2 secretion after 72 hrs of culture, the reduction was not observed when CD4+FoxP3+ iTRegs were fully developed after 120 hrs. Several studies have suggested that iTRegs mediate their suppressive function through the secretion of IL-10 and/or IL-35. However, the role of IL-10 in CD4+ iTReg suppression function has been controversial and conflicting data has been reported both for humans and in mice [51]. Here the differentiation of human CD4+FoxP3+ iTRegs was associated with a significant reduction of IL-10 production and negligible amounts of IL-35 were present in all conditions tested. Cytokine dependent mechanisms have been strongly associated with the suppressive function of iTRegs [52]. Additionally, our findings suggest that such cytokine dependent mechanisms are the driving force of their suppressive function.
Our study indicates that low levels of TNF? and IL-1? promote the induction of CD4+FoxP3+ iTRegs similar to the low intensity and tolerogenic immune response at mucosal sites. However, during high intensity and prolonged T cell activation involving the danger signals of the innate immunity, TReg induction and their function would be suppressed, a condition which would be expected during persistent infection or chronic autoimmune disorders.
Acknowledgements
We thank Dr. Helgi Valdimarsson for his critical comments and reading of this manuscript. Furthermore, we thank Research Fund Landspitali University Hospital, University of Iceland Research Fund and Scandinavian Society for Immunology (SSI) for funding.
Disclosures
The authors declare no conflict of interest.