David J. Ramsey, MD, PhD, MPH*1, and G.B. Arden, B.Sc, MBBS, FRC Ophthal, PhD2
1Department of Ophthalmology, Lahey Hospital & Medical Center, Tufts University Medical School, 41 Mall Road, Burlington, MA, 01805, USA
2Professor Emeritus University College London UK
Corresponding author
David J. Ramsey, MD, PhD, MPH, Department of Ophthalmology, Lahey Hospital & Medical Center, Tufts University Medical School, 41 Mall Road, Burlington, MA, 01805, USA, E-mail: David.J.Ramsey@lahey.org
Received Date: 15th September 2014
Accepted Date: 29th October 2014
Published Date: 10th January 2015
Citation
Ramsey DJ, Arden GB (2015) Rods, Dark Adaptation, and the Development of Diabetic Retinopathy. Enliven: Clin Ophthalmol Res 1(1): 001.
Copyright
@2015 Dr. David J. Ramsey. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
There is a pandemic of diabetes. More than 350 million people are affected world-wide. In the UK more than 4.2 million people (6.3% of the population) are estimated to be living with diabetes, many without even knowing it. In the USA 29.1 million ( 9.3% of the population ) have diabetes. Most individuals have type II diabetes the onset of which is strongly attributed to the adoption of a “western diet” rich in calories from refined carbohydrates and saturated fat. But type I diabetes is also increasing. Furthermore, type II now appears at much earlier ages, even in children, and increasing numbers of people with type II are insulin dependent. Even with an estimated 150,000 people in the UK developing diabetes each year, the prevalence of very long standing diabetes is growing even more rapidly as individuals with diabetes are living longer than ever before. This sets up a perfect storm for complications of diabetes which develop after long periods.
There is a pandemic of diabetes. More than 350 million people are affected world-wide. In the UK more than 4.2 million people (6.3% of the population) are estimated to be living with diabetes, many without even knowing it [1]. In the USA 29.1 million ( 9.3% of the population ) have diabetes [2]. Most individuals have type II diabetes the onset of which is strongly attributed to the adoption of a ?western diet? rich in calories from refined carbohydrates and saturated fat. But type I diabetes is also increasing. Furthermore, type II now appears at much earlier ages, even in children, and increasing numbers of people with type II are insulin dependent. Even with an estimated 150,000 people in the UK developing diabetes each year, the prevalence of very long standing diabetes is growing even more rapidly as individuals with diabetes are living longer than ever before. This sets up a perfect storm for complications of diabetes which develop after long periods.
There are now various ways of controlling diabetes, but sadly these are not as successful in treating the complications of diabetes that are related to the heart, major blood vessels, peripheral nerves, kidneys and the eye. This paper specifically concentratesonthe damage that occurs at the level of the light-sensitive retina at the back of the eye. Already 240,000 people in the UK with diabetes have diabetic retinopathy. When it affects the central few degrees of vision, it causes visual impairment. This may occur from bleeding in the eye, often with accompanying abnormal proliferating blood vessels, retinal detachment from associated scar tissue, or even more commonly the leakage of fluid from damaged capillaries causing diabetic macular oedema (DMO). In the USA DMO is the leading cause of blindness in people of working age [3]. In the UK although the prevalence is similar, there has been a recent increase in the incidence of hereditary retinal degeneration, so DMO is now the second most common cause of registered blindness in the working age-group [4] .
Once diabetes has damaged the retina, vision is often permanently lost. For this reason all efforts are being made to detect diabetic retinopathy as early as possible. Therefore in several countries with national healthcare systems there are National Diabetic RetinopathyScreening services which annually take pictures of the back of the eye. In the UK, for example, over 3 million people are examined in this way, and over 100,000 of these are referred to the Eye Departments of NHS Trusts, which are struggling to cope with these large numbers of patients. (Figure 1) shows such a view taken from the eye of a patient with relatively early disease, mild non-proliferative diabetic retinopathy. The changes clinically apparent are microaneurysms and exudates with an associated thickening of central retina [5]. Microaneurysms are thought to be due in part to loss of pericytes [5]. In the resulting capillary dilatation, blood flow is sluggish and localised clotting may occur. In the end stages of this process the whole capillary is often obliterated. This can be seen histologically as ?ghost vessels? and can be seen on fluorescein angiography of the retinal circulation as localized areas where the inner retinal blood supply is lost. Until a few years ago, the only treatment for diabetic retinopathy was laser photo-coagulation (burns) of the retina. (Figure 2) shows the end results of such treatment. The image is a collage of different areas of retina, photographed with the same equipment as the previous slide. Care is taken to avoid burns in the central region or macula, as this would damage the central vision, although focal laser applications are sometimes used by ophthalmologist within the macula to treat areas of leakage.
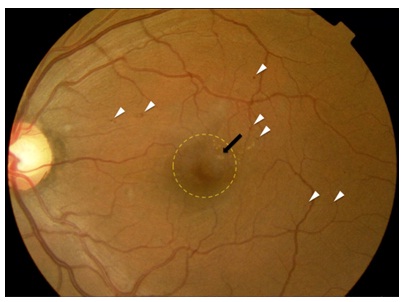
Figure 1: Fundus photograph of a person with mild (early) nonproliferative diabetic retinopathy with centre-involving diabetic macular oedema (DMO) in the left eye. The white arrow-heads highlight typical examples of small red ?dot? (microaneurysms) and ?blot? (haemorrhages). The black arrow points to retinal exudates several of which are within 500 microns of the centre of the fovea. The yellow circle delineates an area of central thickening of the retina (oedema).

Figure 2: A montage of 7 fundus photographs from a patient who had undergone pan-retinal photocoagulation some years previously. The optic nerve head is at the centre, and the blood vessels can be seen radiatingfrom it.
The white spots are scars left from the laser burns. Initially 400 ?M across, they have enlarged with time. In these areas there is no retinal function.
The white areas are fullthickness scars, in which the retina is dead. At first it was thought that the laser treatment prevented injurious substances leaking from those parts of the retina affected by diabetes [6,7], but in fact, the burns can be placed anywhere in the peripheral retina and the treatment is successful in arresting progression of the disease as long as a sufficient area of the retina is treated. But the outcome is far from perfect. The treatment is often given after loss of visual acuity has already occurred, and it does not act to restore vision. Indeed after 5 years, slight further deterioration of acuity is the rule, although vision is conserved in patients that receive treatment compared to those that forgo pan retinal laser photocoagulation [8]. Furthermore, the destruction of the peripheral retina has as an unavoidable side effect loss of peripheral vision and patients often complain of decreased night vision in many cases.
The situation was transformed over the last decade with the introduction of intravitreal agents that target the vascular endothelial growth factor (VEGF), one of the key cytokines whose abnormal expression has been strongly associated with the progression of diabetic eye disease [9]. While awaiting FDA approval for ranibizumab (Lucentis), a humanized small antibody fragment (Fab) with a high binding affinity for human VEGF, which was specifically designed for intraocular use with a smaller size compared to its parent molecule bevacizumab (Avastin) [10], ophthalmologists began treating neovascular AMD with off-label bevacizumab [11] which has since become a widely adopted, lower-cost alternative to ranibizumab. A new drug, aflibercept (Eylea), which targets all VEGF isoforms and PlGF, has all been recently approved for the treatment of DMO [12]. Ongoing studies indicate that these and related compounds may actually slow the progression of DR [13]. Furthermore, they are highly effective therapies for DMO, often resulting in an increase in visual acuity, although some patients do not respond and other have such extensive damage that vision is permanently lost [14-20]. However, it is also unfortunate that they are very expensive [20]. So the list of available treatments is short, and the results and access to treatment are not particularly inspiring. But the important message of this paper is that it is almost certain that an entirely new method of treatment, which is not only non-invasive and without side effects, but is also so inexpensive, and simple you can ?do it yourself? has been discovered. Below the rationale and the supporting evidence is given.
The Hypothesis
Hypoxia, driven by dark adaptation, accelerates the development of diabetic retinopathy [21-23] There are several reasons why this occurs: Rod photoreceptors in the outer retina are maximally depolarized in the dark and continuously release large amounts of theirneurotransmitter glutamate and this increases energy utilization and oxygenconsumption [24,25] (Figure 3 and Figure 4).
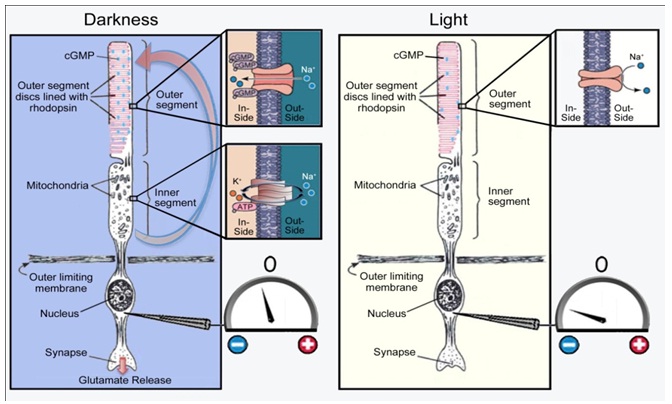
Figure 3: Mechanism of rod excitation. In darkness, non?specific cation channels in the outer segment membrane are open (as indicated by the inset), and there is a net inward gradient of ions and water. This depolarizes the rod, as indicated in the diagram by the voltmeter recording from the nuclear region. In light, the ion channels close and the rod repolarizes (hyperpolarizes). The water and ions entering the rod outer segment are pumped out by pumps localized in proximity to the large mitochondria of the inner segment. This causes a longitudinal current to flow from the inner to outer limb of the cellas indicated by the red arrow. In light this stops. The electrochemical changes that occur in response to an incremental change in light can also be measured electrophysiologically by the flash electroretinogram (ERG), recorded as a negative potential at the cornea known as the a-wave (not shown). modified from [26]

Figure 4: Volume-for volume, the rod in darkness pumps 10 times the volume of a fireboat. This produces heat, and demands a great deal of energy in the form of ATP, primarily produced by oxidative phosphorylation by the large mitochondria of the inner segment of the photoreceptor.
In darkness rods consume more oxygen than any other cell in the body [27,28], and even though the choroid provides a huge circulatory volume, the partial pressure of oxygen in the innersegment layer is near zero compared with the 30 mmHg common in most tissues. In animal experiments, the partial pressure of oxygen rises suddenly and briefly when the retina is stimulated bya flash of light [29]. With normal oxygen concentrations, the rods receive sufficient oxygen, but if this is reduced by breathing reduced gas mixtures, the rod sensitivity falls. This occurs with only a modest reduction of oxygen corresponding to a reduction in air pressure corresponding to an elevation of about 4000 ft. Thus, in commercial jet aircraft, which are pressurised to around 7000 ft., healthy normal observers have slightly reduced rod sensitivity and some individuals, particularly those with heart or lung disease, may experience symptoms of dimmed vision, fatigue, nausea, headaches, loss of consciousness, and (on extended flights) even pulmonary oedema or death from altitude sickness [30]. Consequently, if in retinal disease the availability of oxygen is reduced due to decreased function of the retinal circulation, or the relative demand for oxygen increasesdue to metabolic stress, there will be an imbalance which will lead to hypoxia in not only the outer retina (which is avascular) but also in the inner part, which is supplied primarily,but not completely, by the retinal arterial circulation. These processes are diagrammed in (Figure5).
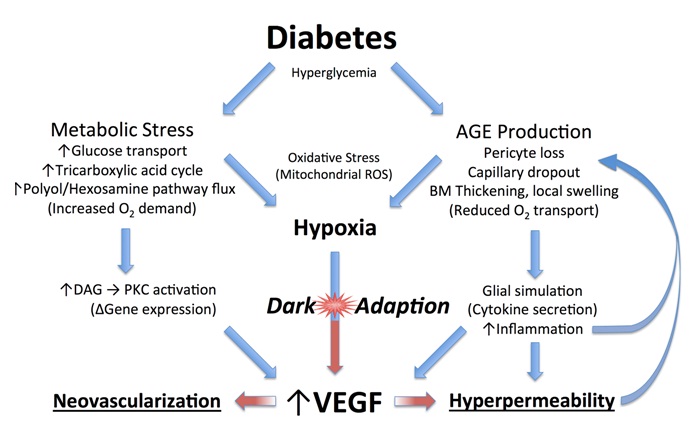
Figure 5: Diagram of how diabetes leads to vascular changes in the inner retina.
Increased blood sugar levels in diabetics will lead to increased glucose transport through cell membranes in many cell types but not apparently in rods due to their high affinity glucose transporters [31,32]. On entering the tricarboxylic cycle, the excess sugars will lead to an increased demand for oxygen. This also results in the formation of advanced glycation end products (AGEs), which are modifications of proteins or lipids that become non-enzymatically glycated when exposed to aldose sugars and oxidative stress [33-35]. Recent experimental evidence shows that rod photoreceptor cells are major contributors to diabetes-induced oxidative stress by producing reactive oxygen species and local inflammation in the retina [36]. These changes accumulate with time affecting both intracellular and extracellular function and by engaging the receptor for advanced glycation end products (RAGE). Activation of RAGE by AGEs causes upregulation of the transcription factor nuclear factor? ?(NF??) and its target genes. In Muller cells this promotes the expression of glial fibrillary acidic protein (GFAP) which is seen very early in diabetes, and the secretion of the cytokine, VEGF. As already alluded to above, VEGF causes changes to small vessels and makes them leaky (VEGF was initially identified as a vascular permeability factor [VPF] [37]). Other AGEs contribute to the increase in basal membrane thickness which occurs in many tissues, including the capillaries of the retinal circulation [38-42].
All these factors reduce oxygen transport into the retina. Additional biochemical alterations are known to occur in the diabetic retina, but once hypoxia is established, it will cause a cascade of effects that drive the production of VEGF leading to further changes associated with diabetic retinopathy that exacerbate any underlying hypoxia [43-47]. Since a major cause of retinal hypoxia is the demand of the rods during periods of dark adaptation, reducing rod dark current should ameliorate diabetic changes. However, many other biochemical changes seen in diabetic retina have been connected to the over production of reactive oxygen species [43-47]. If these pathways are more important, increasing the retinal oxygen tension could either have little effect, or might even accelerate the progression of diabetic retinopathy. However, given that pan-retinal laser photocoagulation, the mainstay of treatment for proliferative diabetic retinopathy, has been shown experimentally to restore local oxygen levels to normal within weeks of its application [48] and is thought to work mainly by destroying large numbers of rods in the peripheral retina there by increasing the oxygen supply to the remaining tissue [49], the benefits of simply preventing rod activity during dark adaptation by the application of constant nocturnal light seem promising. Furthermore, vascular responsiveness to oxygen inhalation also improves following photocoagulation [50-52].
Evidence for the Hypothesis
The hypothesis explains the success of panretinal photocoagulation: the treatment destroys rod photoreceptors, thereby reducing oxygen demand of the outer retina, particularly under dark adapted conditions. The remaining rods benefit, but even more important is the improved ability of oxygen to reach the inner retina from the choroid by virtue of the reduction in rod photoreceptors with their oxygen-absorbing mitochondria. This may change the balance in the supply of oxygen to the inner retina, which has a compromised retinal circulation thereby relieving the conditions which give rise to the overproduction of VEGF.
The use of anti-VEGF drugs is now the most common initial treatment for diabetic eye disease, and hypoxia is the major determinant VEGF production, but this is almost a circular argument. Direct measurement of oxygen tension in diabetic cat retina shows that the inner retina is more hypoxic than that of normal felines even in regions where there is no capillary dropout [53,54] (Figure6).
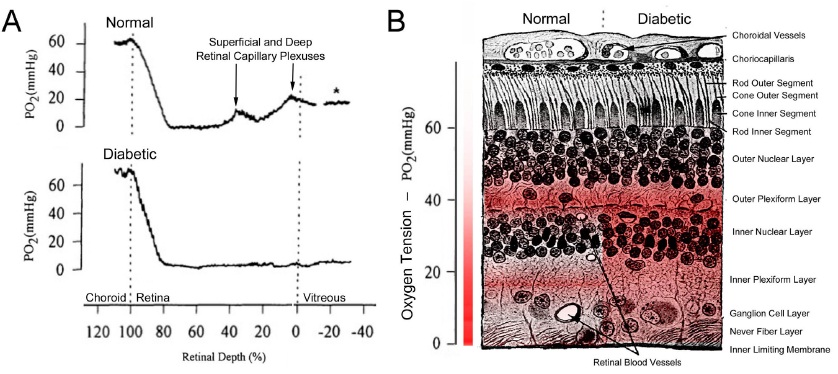
Figure 6: Retinal Oxygen Tension in Normal and Diabetic Retina. (A) Measurements (averaged) of oxygen partial pressure in retina, using O2 microelectrodes. The high tension in the choroid indicates blood with arterial saturation. A: upper part in normal animals the rapid decrease as the electrode is withdrawn (proceed proceeding from left to the right) is followed by an increase as the electrode passes through the deep and superficial capillary plexuses until the vitreous is reached (see * on trace). In the diabetic eye, (lower part) oxygen partial pressure is markedly lower in the inner retina. (Modified after [54]). (B) Cross section of the human retina with the gradient of oxygen partial pressure overlaid in red. Relatively highly colored regions are more oxygen rich. The left hand side of the retina in part B illustrates the conditions present in the normal retina (corresponding to the top trace in part A), whereas the right hand side labeled diabetic illustrates what happens to oxygen tension in the diabetic state (corresponding to the bottom trace in part A). (Modified after [55]).
This is very strong evidence in favour of the hypothesis. Similar but more indirect results have been reported using fMRI that demonstrate that retinal hypoxia in mice increases as the animals dark adapt [56]. The role of rods in diabetic retinopathy is an essential part of the hypothesis-not least because the retina but not the brain (a similar tissue) does not develop such vascular changes [57].
Human beings have many diseases that selectively destroy rods, producing retinal dystrophies-Retinitis Pigmentosa is the most common of these conditions. Rod loss in many cases is slow, and diabetes can occur independently in conjunction with the slow loss of photoreceptors. In such patients, retinopathy is rarely seen (Table 1) [58].
| Classification of diabetes | Type I | Type II | |
| Number of replies validated by doctor | 25 | 30 | |
| Mean age (SD) | 60.8 (16.8) | 65.5 (15.3) | |
| Mean age at diagnosis | 35 | 34 | |
| Mean duration of diabetes | 19 | 14 | |
| Interval between onset of night blindness and diabetes | 7.2 (n=10) | 19.5 (n=24) | |
|
0 | 0 | |
| # of cases of other major diabetic complications (non-ocular) | 5 (n=11) | 8 (n=18) |
Table 1: Incidence of diabetic retinopathy in patients with both diabetes and rod degenerations (retinitis pigmentosa): None of the patients (discovered by a web-based search of patient-societies with surveys validated by attending ophthalmologists) has the characteristic changes, though many had severe vascular and other disease in other organs attributable to diabetes.
Another line of evidence comes from patients with maternally inherited diabetes and deafness (MIDD). These patients have mutation of mitochondrial DNA which manifests in a number of ways and can cause a type of retinal degeneration. This may or may not occur together with diabetes. If the retinal degeneration occurs together with the diabetes, the incidence of micro aneurysms is less than if the diabetes occurs alone [59,60]. In some individuals, the efficiency of hypoxia in increasing VEGF and VEGF RNA levels is much reduced and can be seen by incubating white blood cells under normoxic and hypoxic conditions. Among this group, retinopathy does not occur in individuals who also have diabetes [61]. In addition, breathing oxygen improves several visual functions in patients with diabetic retinopathy and DMO [62-65].
Finally, patients with diabetes who have sleep apnoea have more rapid development of diabetic eye complications than those who do not have sleep apnoea. The increased risk is reduced in patients with sleep apnoea who are treated with positive pressure ventilation [66-70]. There are several recent reviews that include a more detailed discussion of the hypothesis and the evidence for it [71,72].
However, the most convincing evidence has come from clinical trials, in which patients wore ?light masks? which provided retinal illumination through the closed eyelids during sleep. Sufficient light penetrates the lid to partially light adapt the rods. The light sources were either chemo-luminescent (glow-patches) or light-emitting diodes (LEDs). The green or blue-green light was used to selectively stimulate rods (95% of all photoreceptors are rods) rather than cones, because the sensations aroused by rod stimulation are less intrusive and tend to fade to insignificance as patients adapt to a low background luminance. In order to reduce inter-personal differences, the subjects wore masks which illuminated only one eye, with the other, in darkness being used as a control [72]. The results of a 6 month Phase I trial showed that no adverse reactions were encountered and patients accepted the idea of wearing a mask during sleep. However, unusual for a Phase 1 trial, the therapy caused a significant improvement in the 10trial eyes relative to their controls. (Figure 7) shows such an illustrative result.
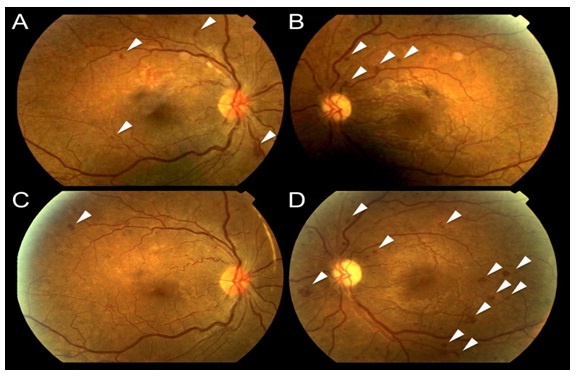
Figure 7: Representative fundus photographs from a patient with diabetic retinopathy. The upper row shows fundus photographs at baseline in the right (A) and left (B) eyes. The lower row shows the same patient following three months of light at night treatment of the right eye (C) with the left eye serving as a control (D). Note that in C the areas of haemorrhage (white arrows) have faded compared to A, consistent with an improvement in the clinical stage of retinopathy. In fact, in many light treated patients no new lesions developed. In contrast, the control eye (B and D) demonstrates many new and worsening haemorrhages at 3 months.
Fundus photographs such as shown in (figure 7) were examined under high magnification and the total areas of micro aneurysms measured. The improvement in the condition (decrease in areas of affected central retina) was significant. A similar result was obtained when changes in tritan ? blue-yellow - colour vision (known to be damaged early in diabetic retinopathy) was assessed (not shown) [73]. In a Phase II trial of 42 patients with non-centre involving macular oedema, a similar benefit was shown. A high proportion of the treated eyes improved, while this did not occur in untreated eyes (Table 2). Furthermore, whereas four of the patients recruited to the trail dropped out because the unilluminated fellow eyes received laser or intravitreal bevacizumab for progression of DMO during the study, none of the illuminated eyes required such treatment.
| Trial Eye | Control Eye | P value* (chi-squared) | |
| Number improved | 17 | 3 | |
| Number unchanged | 6 | 17 | 0.0003 |
| Number worse | 3 | 6 |
Table 2: Change in macular oedema at 3 months. Initial clinical results looking at change in macular thickness in a Phase II trial demonstrated that a majority of eyes treated with light at night improved, while untreated eyes in the fellow patient more often stayed the same or even worsened over the course of the trial [74].
Ocular coherence tomography showed that cysts characteristic of DMO disappeared, and significant reduction in thickness occurred (figure 9 and figure10) [74].
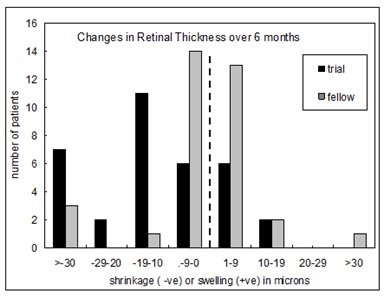
Figure 9: Changes in retinal thickness associated with light adaptation in the Phase II trial [74].
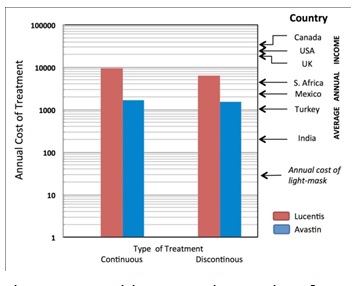
Figure 10: Health Economics results of treatment of DMO. Relative costs (£) of treatment by Lucentis and Avastin used at fixed intervals (continuous) or when required (discontinuous). Note the logarithmic ordinates. Lucentis is 40 times more costly to purchase than Avastin but the difference in the total cost of treatment is much less. At the right, the median incomes in a number of different countries is indicated. Note the only in the most affluent countries is the use of these drugs even possible for most of the population at risk. The cost of light masks is estimated to be far less.
The patients in this trial had very early DMO, and in many patients the swelling did not involve the central EDTRS area within 300 ?M of the fovea. Hence if there is a reduction observed in local swelling in one retinal region might swelling simply be occurring in another region? In order to evaluate this, measurements were made in the ETDRS zones, and the region with maximal swelling was compared to its opposite ?mirror ?zone (Table 3, Figure 8).
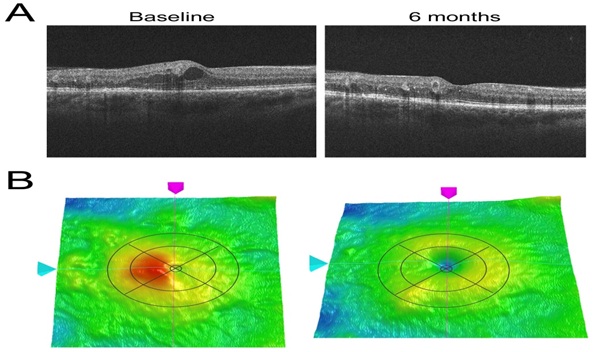
Figure 8: (A) Illustrative OCT results on an eye before and after light treatment Cross-sectional views of the retina showing swelling and cyst at baseline, which has largely vanished at 6 months. (B) The False colour 3-dimensional OCT images from the same examinations. The overlay represents the EDTRS zones. The central small disc is zone 1 and contains the foveal pit. The area of maximal swelling (red) is in this case zone 2 The remaining zones of the inner ring, going clockwise are zones 3,4 and 5, and the outer ring comprises zones 6,7,8,9. The red-false colour in zone 2 indicates an abnormality. In the clinical trial the thickness of the maximally affected zone was compared to the relatively unaffected ?mirror zone? 4 ? or in other instances, if zone 3 was maximally affected, it was compared to zone 5. In this way it could be shown that the oedema was reduced and had not merely moved to a different retinal locus.
| OCT measure of retinal thickness | ||||
| [change in trial eye] ? [ change in fellow eye] (µm) | ||||
| Zone | Mean | SE | 95% CI | P = |
| Central | -11 | 1.92 | -14.7?-6.9 | 0.026 |
| Maximal | -13 | 2.26 | -17.5?-7.2 | 0.052 |
| Mirror | 2.4 | 2.41 | -2.5?7.3 | 0.144 |
Table 3: Summary of changes in OCT measures. The zones are illustrated by the overlays of the OCT images in (Figure 8).
It can be seen that decrease in thickness occurs only in treated eyes. A small but significant improvement in visual acuity and contrast sensitivity also occurred among the light adapted eyes in this trial (Figure 9, Table 4).
| Test | Trail eyes ? numbers of letters read | Fellow eyes ? numbers of letters read | Change and 95% CI after 6 months | |||
| Initial Values | Final values | Initial values | Final values | |||
| Mean SD | Mean SD | Mean SD | Mean SD | |||
| Visual Acuity (ETDRS charts) |
78.04 7.80 Range 85-60 |
80.18 5.32 Range 85-60 |
76.52 8.10 Range 86-67 |
74.59 8.52 Range 87-47 |
4.21 2.64-5.78 |
|
| Constrast sensitivity (pelli - robson) | 31.71 5.39 Range 38-20 |
33.14 4.57 Range 41-20 |
30.16 4.71 Range 36-23 |
30.50 4.95 Range 36-20 |
2 1.25-2.75 |
|
Table 4: Summary of results of visual acuity and contrast sensitivity testing in study and fellow eyes in a Phase II trial of light adaptation [74].
Such studies have provided ?Proof of Principle? that retinal hypoxia in darkness must be an important driving force for diabetic retinopathy. However, further work is required before such treatment could be recommended. We do not know how ?light at night? interacts with the other treatments for the condition. For this Phase III and IV clinical trials are required. This presents a difficulty, because for new pharmaceuticals, such trials are largely funded directly by the drug companies, while there is no such equivalent for medical devices. Small commercial companies have manufactured masks which can be used in such trials, and apparently in one trial carried out in the Department of Ophthalmology, Vinohrady, Prague Czech republic, produced encouraging results which have not been published in detail. Further trials are starting in London, Cardiff (UK), Boston (USA) and the EU and health economic data is being assembled. There is a considerable need for this research because the cost of the present day ?gold standard? treatment is becoming unsustainable [75].
Anti-VEGF drugs are expensive: Avastin costs about 1/40th of the price of Lucentis, but the cost of treatment by the two is not so much different, in part because of the cost of administration (Figure 10). But it is only in the first world where incomes are high enough for a significant proportion of the population with vision threatening diabetic eye disease to access treatment [75]. World-wide there is not only a shortage of funding for available treatments, but a severe shortage of personnel trained to diagnose the condition and manage it appropriately. By contrast, the requirements of light-based treatment to interrupt nocturnal dark adaptation, for example through the use of a light mask as has been used in clinical trials, are relatively inexpensive, and so safe and easy to use that it could be made available with only basic instruction provided by paramedical personnel. This treatment may not replace medical intervention, but promises to complement or reduce the need for traditional therapies thereby reducing the cost of caring for patients with diabetic eye disease. But further research and development is needed. Only then will light masks or a similar device be manufactured on a scale capable of reducing their cost much below the price of Lucentis.
References
3) Facts About Diabetic Retinopathy. National Eye Institute.
5) Joussen AM, Gardner TW, Kirchoff B, Ryan SJ (2007) Retinal Vascular disease. Springer 778.
41) Zong H1, Ward M, Stitt AW (2011) AGEs, RAGE, and diabetic retinopathy.Curr Diab Rep 11: 244-252.
42) Stitt AW (2010) AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci 51: 4867-4874.
55) Minot CS (1907) The Problem of Age, Growth and Death.The Popular Science Monthly. 71: 97-120.