Research Article
John R. Richards, MD1*, Robert W. Derlet, MD1, Timothy E. Albertson, MD, MPH, PhD1,2,3 B. Zane Horowitz, MD4, and Richard A. Lange, MD, MBA5
1Department of Emergency Medicine, Divisions of Toxicology, Pulmonary and Critical Care, University of California Davis Medical Center, Sacramento, CA
2Department of Internal Medicine, Divisions of Toxicology, Pulmonary and Critical Care, University of California Davis Medical Center, Sacramento, CA
3Northern California VA Medical System, Divisions of Toxicology, Pulmonary and Critical Care, University of California Davis Medical Center, Sacramento, CA
4Department of Emergency Medicine, Oregon Health Sciences University, Medical Director, Oregon Poison Center Portland, OR
5Department of Medicine, Division of Cardiology, University of Texas Health Sciences Center, San Antonio, TX
Corresponding author
John R. Richards, MD, Department of Emergency Medicine, PSSB 2100, U.C. Davis Medical Center, 2315 Stockton Boulevard, Sacramento, CA 95817, Tel: (916) 734-1537; Fax: (916) 734-7950; E-mail: jrrichards@ucdavis.edu
Received Date: 10th July 2014
Accepted Date: 21st August 2014
Published Date: 26th August 2014
Citation
Richards JR, Derlet RW, Albertson TE, Horowitz BZ, Lange RA (2014) Methamphetamine, ?Bath Salts,? and other Amphetamine-Related Derivatives: Progressive Treatment Update. Enliven: Toxicol Allied Clin Pharmacol 1(1): 001.
Copyright
@ 2014 Dr. John R. Richards. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, that permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.reproduction in any medium, provided the original author and source are credited.
Abstract
Background: The abuse and accidental overdose of both prescribed and illicit amphetamine and related compounds such as methamphetamine, “bath salts,” methylphenidate, cathinone (“khat”), and MDMA (“ecstasy”) is a worldwide problem affecting millions. Patients experiencing toxicity from these drugs frequently present to the emergency department and may have serious consequences from their hyperadrenergic state, such as acute coronary syndrome, stroke, acute heart and renal failure.
Objective of the Review: Medical management of these patients has changed over time as new therapies have been developed, with control of agitation and hyperadrenergic state being the top priorities to prevent secondary injuries.
Discussion: Control of agitation with benzodiazepines and antipsychotics is an important first step but may not alleviate the tachycardia, hypertension, and hyperthermia these patients often experience. Unlike cocaine, the half-lives of these drugs are several hours with increased potential for pathologic sequelae. The risk of adverse events may not be linear with the long half-lives of these compounds. This requires the treating clinician to be flexible to different therapeutics and adjust management accordingly.
Conclusion: In this comprehensive review, the pharmacology, pathophysiology, and level of evidence behind treatment of patients with toxicity from amphetamine, its analogues, and related derivatives are discussed and recommendations given. The development of potential new therapeutics and future research direction are also discussed.
Introduction
Abuse of amphetamine, its analogues, and related derivatives (AAD) is a growing worldwide problem. In 1989 Derlet and Horowitz published the first large case series of 127 patients with amphetamine toxicity in the emergency department (ED) [1]. The problem of AAD abuse continued to grow during the next several years. In 1999 Albertson and Derlet reported myriad serious medical complications from AAD abuse and overdose [2]. In the United States, there were an estimated 159,840 ED visits for toxicity from illicit use of amphetamines in 2011 [3]. Exposure to ?bath salts? has increased exponentially in the United States from 2009 to 2011 according to data from the National Poison Data System [4]. Patients who are first-time or chronic users of AAD, which may include (Figure 1) methamphetamine,3,4-methylenedioxy-N-methylamphetamine (MDMA, ?ecstasy?), cathinone (khat), and methylenedioxypyrovalerone (MDPV) or mephedrone (?bath salts?) frequently present to the ED for diverse reasons [5]. Death from AAD may occur before the patient can receive emergency care. Many have underlying psychiatric disorders such as schizophrenia, bipolar disorder, and personality disorders which may confound their clinical presentation after the use of AAD [6-10]. Abuse of prescribed AAD, such as amphetamine/dextroamphetamine (Adderall®), lisdexamfetamine dimesylate (Vyvanase®),and methylphenidate (Ritalin®) for conditions such as narcolepsy and attention deficit hyperactivity disorder (ADHD) among high-school and college students is a growing problem [11-13].
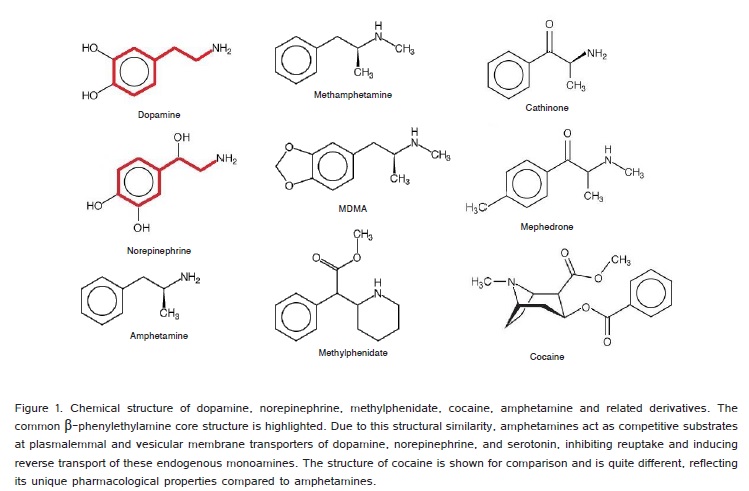
In this review these drugs are grouped as AAD, as there is often uncertainty of which drug is involved, and these patients are rarely forthcoming about their illicit drug use. This class of drugs also includes over-the-counter decongestants and anorexiants such as ephedrine, pseudoephedrine, and phenylpropanolamine. Until results of a urine drug screen are available or the patient admits to the ingestion, the clinician must consider other etiologies resulting in a hyperadrenergic state, such as agitated psychosis, sepsis, thyrotoxicosis, and pheochromocytoma (Table 1). Qualitative urine toxicology screening tests commonly used in the ED may not identify specific prescribed drugs like methylphenidate, and often are not sensitive enough to pick up diverse illicit amphetamine compounds, such as the ?bath salt? mephedrone derivatives [14].
| SympathomimeticsIllegal: Amphetamines, Cocaine, ?Bath Salts,? PhencyclidineLegal: Beta-agonists, Decongestants. |
| Overdose: Anticholinergics, Antihistamines, Monoamine oxidase inhibitors, Salicylate, |
| Alcohol withdrawal |
| Opioid withdrawal |
| Benzodiazepine withdrawal |
| Acute psychosis |
| Seizures and post-ictal state |
| Sepsis |
| Stroke |
| Acute coronary syndrome |
| Toxemia of pregnancy |
| Acute heart failure |
| Paroxysmal supraventricular tachycardia |
| Thyrotoxicosis and thyroid storm |
| Serotonin syndrome |
| Neuroleptic malignant syndrome |
| Hyperthermia |
| Anxiety/Panic attack |
| Pheochromocytoma and paraganglionoma |
| Carcinoid tumor |
| Autonomic neuropathy (baroreflex failure) |
| Renovascular or labile essential hypertension |
| Subarachnoid hemorrhage |
| Head trauma |
| Hypoglycemia |
| Hypoxemia |
| Tetanus |
Table 1.Differential Diagnoses of the Hyperadrenergic State.
For this review, an extensive and thorough search of the published literature from the earliest possible date to April 2014 regarding toxicity and treatment of these drugs was performed in MEDLINE®, revealing over 1,000 animal and human studies, case reports, reviews, and scientific investigations. References in each of these publications were also carefully screened for any additional reports which may have relevance to this review. Particular attention was given in determining any adverse outcome associated with each specific treatment.
Discussion
Pharmacology and Effects
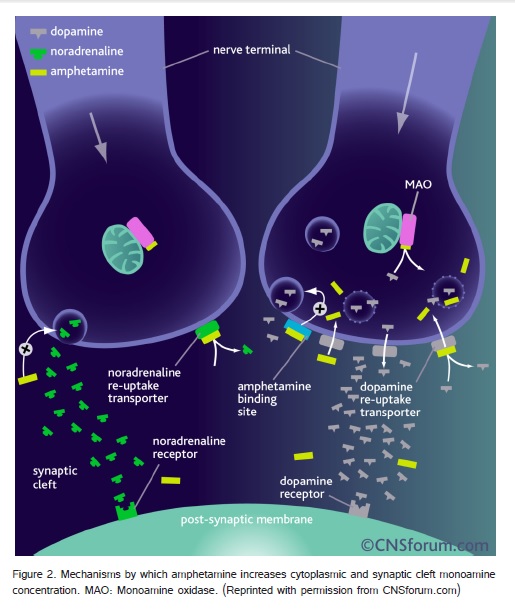
Amphetamine, its analogues, and related derivatives are orally ingested, smoked, snorted, or injected. Vaginal and rectal absorption is also possible. The half-life of AAD range from 4-12 hours, compared to approximately one hour for cocaine. Peak plasma levels and subjective effects are observed minutes after intravenous (IV), intramuscular (IM) use [15]. Intranasal or vapor inhalational routes may result in peak subjective effects in minutes, but peak plasma levels occur approximately 2-3 hours later due to the mucosal barrier. Oral ingestion results in peak plasma levels 3-6 hours later. Hepatic conjugation pathways with glucuronide and glycine addition result in inactivation with urine excretion of metabolites. The rate of excretion is increased by acidic urinary pH. These drugs increase serum levels of the monamines norepinephrine, dopamine, and serotonin through multiple mechanisms (Figure 2) and are amphipathic molecules which can cross the blood-brain barrier and placenta. Blockade of plasmalemmal and vesicular transporters results in elevated levels of monoamines in the cytoplasm and synapse, respectively [16,17]. With increasing concentration, there is reverse transport of cytoplasmic monoamines across the cell membrane of the presynaptic neuron into the synaptic space, disruption of vesicular monoamine storage, and inhibition of the degradative enzymes monoamine oxidase A and B.
Agitation and Hyperthermia
The psychostimulant effects of AAD are mediated primarily through dopaminergic neurons within the central nervous system (CNS). Low-to-moderate doses increase alertness, attention, and concentration, especially in sleep-deprived users [9]. Other effects include decreased appetite and elevated confidence, mood, and libido. Higher doses cause agitation, restlessness, anxiety, and dyskinesia. Chronic ?binge? use over a period of days, results in dysphoria, psychosis, paranoia, and compulsive behavior, or ?tweaking? [18]. With higher doses also comes the risk of interpersonal violence, suicidality, and increased frequency of motor vehicle accidents [16]. Patients presenting to the ED with toxicity from AAD are often agitated, uncooperative, mendacious, and have markedly elevated heart and respiratory rate, blood pressure, and in some cases, hyperthermia [8]. This places the patient at risk for serious secondary injuries (Table 2).
| Acute coronary syndrome |
| Tachyarrhythmia |
| Hypertensive emergency |
| Pulmonary hypertension |
| Stroke |
| Cardiomyopathy |
| Arterial dissection |
| Mesenteric ischemia |
| Placental abruption |
| Fetal demise |
| Acute renal failure |
| Rhabdomyolysis |
| Vasculitis |
| Seizure |
| Cellulitis and abscess |
| Necrotizing fasciitis |
| Compartment syndrome |
| HIV, Hepatitis B, C |
| Periodontal disease |
Table 2. Toxicity of Amphetamines: Secondary Injuries and Deleterious Effects
Acute Coronary Syndrome
Amphetamine, its analogues, and related derivatives have many deleterious cardiac effects with a significant incidence rate. Acute coronary syndrome (ACS) associated with AAD was first described by Orzel in 1982 [19,20]. This complication has been described in children and adults with varying coronary artery disease (CAD) risk profiles [21]. Sztajnkrycer and co-workers reported ACS and acute congestive heart failure (CHF) with pulmonary edema from Adderall® overdose in a 13-year-old girl [22-26]. A large-scale epidemiological study revealed a significant association of abuse of these drugs and ACS in patients 18 to 44 years of age after controlling for cocaine, alcohol, tobacco, hypertension, diabetes, lipid disorders, obesity, congenital and coagulation defects [27]. Acute coronary syndrome from abuse of other prescription AAD and ?club drugs? such as MDMA has been described in case reports [28]. This risk is also associated with naturally-occurring compounds such as cathinone from the khat plant (Catha edulis) used in the horn of Africa and Middle East, and ephedrine and pseudoephedrine from Ephedra sinica, which are commonly ingested by inhabitants of East Asia [29,30].
Potential mechanisms of AAD-associated ACS are multifactorial and include epicardial and microvascular coronary artery vasospasm, catecholamine-mediated platelet aggregation and thrombus formation, accelerated atherosclerotic plaque formation and rupture, increased myocardial oxygen demand, and direct myocardial toxicity [31,32]. A predominant mechanism has not been identified [19,20]. Although coronary arteriography has identified CAD and coronary thrombosis in some individuals with ACS following use of AAD, many have no angiographic evidence of CAD. Interestingly, a transient, diffuse ?low-flow? state (i.e., delayed coronary blood flow) in the absence of CAD was documented angiographically in a patient with ST-segment elevation myocardial infarction and cardiogenic shock following methamphetamine use, suggestive of transient microvascular dysfunction [33,35].
Cardiomyopathy
Cardiomyopathy from abuse of AAD has been well-documented. In animal models acute methamphetamine exposure directly affects myocyte contractility by inhibition of intracellular calcium release and upregulation of the sodium-calcium exchanger. Methamphetamine also directly damages myocardium by increasing p53 expression as well as the cardiac Fas- and mitochondria-dependent pathways, leading to apoptosis [36]. These drugs induce oxidative stress from accumulation of reactive oxygen and nitrogen species [37]. This spectrum of deleterious effects is postulated to lead to dilated cardiomyopathy seen in methamphetamine abusers [38]. In addition, chronic elevation of catecholamines leads to cardiac remodeling with subsequent hypertrophy and fibrosis [39,40]. Myonecrosis, mitochondrial derangement, enlargement of sarcoplasmic reticulum, small round cell infiltration, and eosinophilic changes are observed microscopically after chronic methamphetamine abuse [41,42]. Takotsubo cardiomyopathy is defined by transient systolic dysfunction of the apical segments of the left ventricle, also described as apical ballooning [43,44]. Amphetamine, its analogues, and related derivatives have been shown to induce reverse (or inverted) Takotsubo cardiomyopathy, in which the basal and midventricular segments of the left ventricle are akinetic. Severe reversible cardiomyopathy has been reported following use of a ?bath salt? compound containing mephedrone and MDPV [45-47].
Vascular, Renal, and tissue injury
In addition to ACS and cardiomyopathy, acute and chronic abuse of AAD is associated with spontaneous aortic and coronary artery dissection, and ruptured cerebral aneurysms. Ischemic and hemorrhagic cerebrovascular accidents have also been reported as well as aseptic cerebral vasculitis [48-50]. Disseminated intravascular coagulation with subsequent death has been detailed [51-54]. In the lung, AAD increase serotonin concentration from upregulation of tryptophan hydroxylase, serotonin transporters, and down regulation of monoamine oxidase, resulting in pulmonary arterial hypertension [55]. Acute and chronic renal failure from necrotizing renal vasculopathy, rhabdomyolysis, and ischemic colitis are associated with use of these drugs [56,57]. Endocarditis, pericarditis, and sepsis from injection of these drugs has been reported [58-64]. In pregnancy, AAD target placental monamine transporters and are implicated in fetal and maternal death [65]. Severe tissue damage from injection, such as compartment syndrome and necrotizing fasciitis, have been described in several case series [66-68]. The actual incidence of many of these deleterious effects is unknown, as most of these references are case reports and not large scale studies [69-71].
Initial Treatment
To prevent harm to patient and ED staff, rapid treatment of agitation is a top priority. This may even require rapid sequence induction and endotracheal intubation. After their agitation has been controlled, some patients may continue to have tachycardia, hypertension, and hyperthermia for several hours, placing them at risk for the aforementioned life-threatening CNS and cardiovascular events [72].
Benzodiazepines and Antipsychotics
Benzodiazepines such as lorazepam, diazepam, and midazolam are benzodiazepine receptor agonists which enhance the inhibitory effects of ?-aminobutyric acid (GABA). Benzodiazepines represent a first line treatment for generalized agitation in the ED. These agents are widely available in acute care settings, and clinicians are familiar with their use [73]. In a large series of ?bath salt? cases, over half were agitated and tachycardic, and 46% of cases received benzodiazepines. Serial IV doses are typically required to achieve restraint, and at much higher dosing range than for non-agitated individuals [74]. Over-sedation and respiratory depression are a risk of frequent and higher doses of benzodiazepines, while paradoxical agitation is another potential unpredictable adverse effect [75,76].
Antipsychotics are CNS dopaminergic receptor antagonists which are also an important mainstay of treatment for agitation from AAD. One advantage of these drugs is blockade of the increased CNS dopamine levels resulting from AAD toxicity [77-82]. The butyrophenones haloperidol and droperidol, and second generation agents ziprasidone and olanzapine have all been successfully used in this setting. Benzodiazepines and antipsychotics may be used alone or in combination. In the only large scale randomized study to date of treatment of methamphetamine users in the ED, Richards and Derlet compared IV lorazepam to droperidol for control of agitation. Both drugs were effective at controlling agitation, but droperidol resulted in faster time to sedation whereas lorazepam required repeat dosing to achieve sedation [82]. In a double-blind 4-week study of 58 patients with amphetamine psychosis, olanzapine and haloperidol were both effective, but olanzapine had a better safety profile with fewer extrapyramidal symptoms. This study, however, did not involve acute management of patients in the ED [79].
Later studies comparing midazolam with droperidol and ziprasidone had similar results, with midazolam requiring more repeat dosing to achieve effect and in some cases, over-sedation. These studies were not specific to agitation from AAD, however [83-85]. The combination of benzodiazepines with antipsychotics has been shown to be synergistic in one study of agitated patients in the ED, with reduced need for repeat administration of benzodiazepines. A Cochrane review of psychosis-induced agitation concluded that combination therapy did not confer an advantage over benzodiazepines or antipsychotics alone for treatment of agitation [86]. Antipsychotics may result in QT interval prolongation, and telemetry monitoring is recommended for patients receiving these drugs for agitation [73]. The possibility of hyperthermia induction from use of antipsychotics was a potential risk with phenothiazines such as chlorpromazine, due to their anticholinergic properties. Phenothiazines are no longer used for treatment of acute agitation, and the risk of hyperthermia is theoretical and overemphasized with butyrophenones and second generation antipsychotics. In fact, hypothermia appears to be a greater risk with these agents [87,88].
Beta-Blockers
Beta-blockers with lipophilic properties, such as propranolol, metoprolol, and labetalol mitigate agitation via CNS-mediated specific antagonism, and non-specific membrane-stabilizing effects. Hydrophilic ?-blockers modulate autonomic hyperactivity in the peripheral nervous system [89]. Several animal studies of agitated behavioral states induced by AAD and treated with propranolol confirms this effect. There have been no human studies of antagonism of AAD-induced agitation with ?-blockers; however, case reports exist of other states of agitation, such as from head injury and schizophrenia refractory to benzodiazepines and antipsychotics, successfully treated with ?-blockers [90-103].
Ketamine and Propofol
Ketamine, a N-methyl-D-aspartate (NMDA) receptor antagonist, has been successfully utilized for control of generalized agitation in a small number of reports. Ketamine produces dissociation and a trancelike cataleptic state while protecting airway reflexes and respiratory drive [104-106]. Difficulties in using ketamine in this situation include emergence agitation and catecholamine surge after administration, leading to a potentially detrimental acute rise in blood pressure, heart rate, and cerebral blood flow. The decreased catecholamine uptake and stimulation of CNS sympathetic outflow from ketamine may be problematic in AAD toxicity, as these patients often have acute hypertension and tachycardia [107]. Propofol is a unique sedative with several mechanisms of action, including GABA receptor agonism, inhibition of NMDA receptors, and alteration of serotonin and endocannabinoid levels. It has been effectively used alone and in combination with ketamine (?ketofol?) for control of generalized agitation in a small number of case reports [108]. Disadvantages of propofol for control of agitation are it requires a continuous infusion by the treating physician, who may be required to remain at the bedside, and it may produce respiratory depression requiring supplemental oxygen and early airway intervention [109-114].
Dexmedetomidine
Dexmedetomidine is a promising agent for control of agitation. It is an ?2-adrenoceptor agonist leading to inhibitory effects on CNS sympathetic outflow, producing sedation, analgesia, and no respiratory depression [115]. Dexmedetomidine has the added benefit of sympatholysis to counteract the cardiovascular and central nervous system overstimulation from AAD. Based on several case reports, dexmedetomidine has been successfully used to control agitation in adult and pediatric patients with toxicity from AAD and cocaine. Other agitated states in which it has been used include alcohol withdrawal and post-electroconvulsive therapy agitation [116-122]. In comparison to haloperidol for control of agitation in delirious, agitated, intubated intensive care unit (ICU) patients, dexmedetomidine was superior with regard to time to extubation, length of stay, and complication rate [123,124]. The main disadvantage of dexmedetomidine is its current cost and limited availability in the ED [125].
Detoxification
There is no specific antidote for AAD toxicity. Patients who are body-packers, body-stuffers, or ?parachuters? may require whole bowel irrigation with polyethylene glycol solution to accelerate transit of drug-containing packets. Detoxification with activated charcoal may be considered for patients arriving to the ED shortly after suspected or confirmed oral ingestion, but this benefit has only been proven in an animal study [126,127]. Both whole bowel irrigation and activated charcoal treatment require a patient who is either intubated or cooperative and alert [128]. The risk of aspiration in a heavily sedated or agitated patient outweighs potential benefit. Hydration with IV crystalloid solution is important as these patients are often dehydrated. Acidification of urine to enhance elimination of AAD has been associated with a higher risk of acute renal failure when rhabdomyolysis is present, and as such, is not recommended care [58,59].
Hyperthermia
The hyperthermia caused by AAD may be due to excessive sympathetic stimulation with skeletal muscle thermogenesis. Agitation and active opposition of physical restraint exacerbate this condition [129]. Hyperthermia necessitates reduction of body temperature, most easily and reliably accomplished in the ED by cool mist application and a fan, application of ice packs and/or cooling blanket, gel packs, and administration of cold IV crystalloid. Cold water immersion is also effective but is precluded when caring for an agitated, sedated, or intubated patient requiring cardiac and pulse oximetry monitoring [130-132]. Careful monitoring of body temperature is important to avoid iatrogenic hypothermia or treatment failure. The hyperthermia induced by MDMA and certain ?bath salts? such as MDPV is believed to be a form of serotonin syndrome. Mugele and colleagues reported a patient who ingested MDPV and developed serotonin syndrome worsened with iatrogenic administration of fentanyl [133]. The patient eventually recovered after receiving several days of cyproheptadine, a first-generation antihistamine with antiserotonergic, anticholinergic, and local anesthetic properties [134]. Dantrolene, a muscle relaxant that depresses excitation-contraction coupling in skeletal muscle, has been used successfully for MDMA-induced hyperthermia. A systematic review revealed improved survival rate in patients with extreme hyperthermia from MDMA who received dantrolene versus those who did not [135]. Hysek et al. showed carvedilol, a mixed ?1/?1-adrenoceptor antagonist, attenuates MDMA-induced hyperthermia and suggested the reduction of temperature was a result of the ?1- and ?3-adrenoceptor blocking properties of carvedilol to modulate peripheral vasoconstriction, heat dissipation, and lipolysis [136,137].
Treatment of Tachycardia and Hypertension
To prevent secondary cardiovascular, renal, and cerebral injury from AAD, treatment of persistent tachycardia and hypertension may be necessary. In addition to the sympatholytic properties of dexmetedomidine, ?-blockers, ?1-adrenoceptor antagonists, nitric oxide-mediated vasodilators, and calcium channel blockers may be used to antagonize the hyperadrenergic state that may persist even after adequate sedation has been achieved.
Beta-Blockers
The majority of published research regarding treatment of toxicity from AAD involves ?-blockers in animal models. In a 1968 study, propranolol, a non-specific ?1/?2-adrenoceptor antagonist which had just become available, effectively antagonized amphetamine-induced hyperthermia and lethality in mice. Subsequent animal studies confirmed the protective effects of propranolol as well as carvedilol [138]. In mice, propranolol blocks in vivo amphetamine-induced norepinephrine release from the heart, presumably from presynaptic ?-adrenoceptor antagonism and prevents methamphetamine cardiomyotoxicity [139-146]. This form of AAD-induced cardiomyopathy in humans has been shown to be reversible with cessation of the inciting drug and ?-blocker therapy [147-153].
There are several human studies and case reports involving ?-blockers and AAD, as well as monoamines such as epinephrine. In a study of healthy adults, Nurnberger et al demonstrated propranolol attenuates IV dextroamphetamine-induced increase in heart rate and systolic blood pressure. Epinephrine infusion in healthy human volunteers results in positive chronotropy and skeletal muscle vasodilation; these effects are blocked by labetalol, a mixed ?1/?2/?1-adrenoceptor antagonist, but reversed by propranolol, suggesting an active role of the ?-blocking property of labetalol [154]. Hassan and associates reported atenolol, a ?1-selective adrenoceptor antagonist, but not indoramin, an ?1-antagonist, lowers systolic blood pressure in khat (cathinone) chewers [155,156].
In a study of human subjects, Hysek et al showed carvedilol attenuated MDMA-induced increase in heart rate and blood pressure. Another study by this research group revealed pindolol, a non-selective ?-blocker with intrinsic sympathomimetic activity, reduced heart rate but not mean arterial pressure after MDMA [137]. From a study of anesthesia patients receiving ephedrine for hypotension who became acutely hypertensive, the authors reported resolution of with labetalol [157]. The interaction of pseudoephedrine and ?-blockers was evaluated in a prospective study in which propranolol and atenolol decreased systolic blood pressure and heart rate [158]. Pentel and co-workers showed propranolol given to normotensive subjects before and after phenylpropanolamine, a decongestant and anorexiant banned in the United States, Canada, and India, decreased blood pressure, cardiac output, and systemic vascular resistance [159,160].
Several case reports are worth discussing. Larsen described a case report of a 31-year-old asthmatic who accidentally received 3 mg subcutaneous epinephrine and developed sudden agitation, diaphoresis, and vomiting. This patient was successfully treated with 5 mg IV labetalol [161]. Pseudoephedrine, at therapeutic dosage induced chest pain, tachycardia, and inferior lead ST-elevation on electrocardiogram in a previously healthy 45-year-old male. Administration of metoprolol IV immediately resolved his chest pain and ST-elevation. Another case report of a 23-year-old male with severe hypertension, headache, and vomiting after ingesting 840 mg of pseudoephedrine revealed resolution of these adverse symptoms with two IV doses of labetalol [162]. Sakuragi and colleagues reported a patient who accidentally received 200 mg IV ephedrine during elective surgery, with subsequent blood pressure of 265/165 mmHg and heart rate 140 beats per minute [163]. Propranolol was administered and successfully mitigated this patient?s hyperdynamic state [164]. A 14-year-old female who overdosed on ephedrine and phenylpropanolamine experienced tachycardia up to 180 beats per minute with arrhythmias including premature atrial contractions, junctional ectopic beats, and short runs of ventricular tachycardia. She failed to respond to lidocaine infusion [165]. Intravenous propranolol rapidly converted her to normal sinus rhythm.
It is recognized that combined ?/?-blockade is advantageous to ?-blockade alone in patients with ischemic heart disease. There are several other hyperdynamic conditions characterized by catecholamine excess, such as cocaine toxicity, ACS, stroke, thyrotoxicosis, pheochromocytoma, paraganglionoma, tetanus, burns, preeclampsia, sepsis, and intense exercise [166-168]. Beta-blockers have been successfully utilized in all these syndromes. It is important to note, however, these conditions may be chronic or involve neurohormonal or physiological pathways not comparable to the hyperdynamic condition induced by AAD [169-184]. Since AAD have been associated with stroke, it is worth noting the 2013 American Heart Association/American Stroke Association guidelines, which recommend labetalol should be initial treatment for hypertension in the early management of acute ischemic stroke. The use of labetalol as an adjunct for treatment of cocaine- and methamphetamine-associated chest pain has been sanctioned by the American College of Cardiology Foundation/American Heart Association [185]. According to the most recent 2012 guidelines for the management of patients with unstable angina and non-ST-elevation myocardial infarction: ?Administration of combined alpha- and beta-blocking agents (e.g., labetalol) may be reasonable for patients after cocaine use with hypertension (systolic blood pressure greater than 150 mm Hg) or those with sinus tachycardia (pulse greater than 100 beats per minute) provided that the patient has received a vasodilator, such as nitroglycerin or a calcium channel blocker, within close temporal proximity (i.e., within the previous hour)? [186].
Unopposed Alpha Stimulation
The concept of ?unopposed ?-stimulation,? with worsening hypertension and/or coronary arterial vasoconstriction after ?-blockade in patients with hyperadrenergic symptoms from cocaine, is controversial. In our extensive literature review, only two small studies and one possible case report of this theoretical adverse outcome in the treatment of toxicity from AAD with ?-blockers could be found [187-190]. O?Connell and Gross reported 7 subjects with hypertension already taking ?-blockers had higher peak blood pressure after single and multiple doses of 25 mg phenylpropanolamine versus placebo. These increases were modest, with peak systolic averaging 8 mm Hg higher than placebo in the single-dose study, and 3-22 mm Hg higher in the multiple dose study [191,192]. The blood pressure measurements were taken on the first and last days of the study period. The authors concluded this effect may reflect the predominant ?- versus ?-mediated properties of phenylpropanolamine in the setting of non-selective ?-or selective ?1-adrenoceptor blockade. The results of this study contradict findings of a similar study using a higher dose of phenylpropanolamine (75 mg) discussed previously. A case report from 1989 was published of a 40-year-old male who inhaled a powder mixture of p-methylamphetamine and N, p-dimethylamphetamine and received IV practolol, a non-specific agent similar to propranolol, for tachycardia of 150 beats per minute and blood pressure 200/120 mm Hg [160]. His pressure rose to 240/160 mm Hg and heart rate fell to 115 beats per minute after practolol, and after several hours normalized without further treatment [193]. This may have occurred from delayed absorption of AAD rather than unopposed ?-stimulation. The authors of the case report concluded labetalol would be a more appropriate choice.
There are two case reports in which ?-blockers were used and putative coronary vasoconstriction subsequently occurred. The first case involves a 37-year old female with chest pain with ST-elevation after IV amphetamine use. Echocardiogram performed the second day after admission showed severe cardiomyopathy and ejection fraction of 33% [194]. Six days after admission, she received two doses of oral propranol and again developed chest pain and ST-elevation that resolved with nitroglycerin. Her coronary catheterization after this second event was normal. It is implausible this represents an adverse stimulant/?-blocker interaction, as amphetamine would have been fully metabolized after 6 days. There is another report of a 19-year old male with who developed chest pain after taking higher than recommended doses of pseudoephedrine. He waited 19 hours before seeking medical attention [195]. His symptoms resolved with nitroglycerin, acetylsalicylic acid, heparin, and atenolol. Several hours after admission his chest pain and ST-elevation returned and resolved with nitroglycerin. His coronary angiogram was normal. As with the previous case, it seems unlikely the recurrence of chest pain was stimulant/?-blocker-related, as there was no temporal association with administration of atenolol and pseudoephedrine should have been significantly metabolized by 24 hours. Finally, neither case report describes a hyperadrenergic state at the time of the recurrence of chest pain.
Alpha-Blockers
The ?1-adrenoceptor stimulatory properties of AAD are variable and not well-defined. There is one human study of ?-blockers for treatment of hyperadrenergic symptoms from AAD and was discussed in the previous section. In a case report from 1969, phentolamine, an ?1-antagonist, successfully resolved a hypertensive emergency induced by phenylpropanolamine [156]. There are two case reports of AAD-induced peripheral arterial vasospasm in which the ?-blockers tolazoline and phenoxybenzamine were used [196]. One treatment was successful and the other failed ?-blocker treatment and required nitroprusside to reverse vasoconstriction [197,198]. Broadley and co-workers have shown the coronary and aortic vasoconstriction caused by cathinone and MDMA is not due to indirect or direct ?1-sympathomimetic activity. A study of MDMA in rats showed pretreatment with phentolamine did not reduce the magnitude of the MDMA pressor response [199,200]. Conversely, in another rat study of mephedrone, Varner and associates demonstrated phentolamine or propranolol mitigated both the pressor response and tachycardia [201]. Yet another study in mice showed phentolamine pretreatment resulted in elevated blood pressure and heart rate and reduced cerebral blood flow after methamphetamine administration [202]. Lange and colleagues showed intracoronary phentolamine reversed cocaine-induced coronary artery vasoconstriction and hypertension, but not heart rate [203]. There are only two case reports of use of phentolamine for cocaine-associated ACS [204]. Despite this rather limited evidence, phentolamine has been recommended by some authors as preferred treatment for cocaine-induced hypertension and ACS [205,206]. The most recent American College of Cardiology Foundation/American Heart Association guidelines for the management of methamphetamine and cocaine patients with unstable angina and non-ST-elevation myocardial infarction do not include phentolamine [186,207,208].
Nitroglycerin and Nitroprusside
There are no studies of the nitric oxide-mediated vasodilators nitroglycerin or nitroprusside for treatment of toxicity from AAD, only cocaine. There are case studies in which nitroglycerin alleviated pseudoephedrine-induced chest pain. Nitroglycerin is helpful in cocaine-induced chest pain and ACS, but does not mitigate tachycardia [209-211]. Lange and associates reported nitroprusside reduced cocaine-induced hypertension, but also increased sympathetic discharge nearly 3 times above baseline [212-215].
Calcium Channel Blockers
Compared to ?-blockers, much less has been published regarding the use of calcium channel blockers for toxicity from AAD both in animal and human subjects. Results of these studies have been inconsistent. In a study of 10 healthy human subjects pretreated with oral diltiazem and then given oral dextroamphetamine, Fabian and Silverstone showed diltiazem significantly prevented rise in blood pressure. In another human study, Johnson and associates reported isradipine, a dihydropyridine-class calcium channel antagonist, reduced methamphetamine-induced rise in blood pressure, but this was offset by reflex rise in heart rate [216]. This same group confirmed this result again in a later study using both cocaine and methamphetamine [217]. Other studies have involved AAD and cocaine or cocaine alone [218]. Schindler et al reported nimodipine, verapamil, and diltiazem antagonized the pressor effect of cocaine but did not mitigate tachycardia in monkeys. Derlet and Albertson reported diltiazem, verapamil, and nifedipine potentiated the seizure and death rate of mice treated with high-dose cocaine [219]. This finding was confirmed in a later study of mice under similar conditions by another research group [220]. Another study showed diltiazem prevented myocardial infarction but not death in mice given a lethal dose of cocaine [221]. Verapamil and nifedipine were shown to reduce the myocardial depressant effect of cocaine with subsequent decrease in ejection fraction in a dog study [222]. Lange and co-workers studied 15 patients undergoing coronary catheterization who received sub-recreational doses of intranasal cocaine [223]. Patients were given saline or verapamil IV, and verapamil decreased blood pressure, coronary arterial vasoconstriction, but not heart rate [224].
Conclusions
Amphetamine, its analogues, and related derivatives have much longer half-lives than cocaine, and thus longer potential for serious complications such as ACS, acute CHF, stroke, hyperthermia, and rhabdomyolysis. Increased widespread use of prescription and illegal AAD has enabled these compounds to be available to the general population. The frequency of abuse and accidental overdose of these drugs will likely continue to rise in the future worldwide. Control of agitation is essential, and the first line therapy of benzodiazepines and/or antipsychotics should be initiated. Sedation with these agents may not attenuate all of the sympathomimetic effects of this class of drugs. Lipophilic ?-blockers and dexmedetomidine are promising agents for both sedation and sympatholysis. Unfortunately, dexmedetomidine is often not readily available in many EDs.
Treatment of tachycardia and hypertension induced by AAD may be necessary to prevent secondary injury. Labetalol has been safely used for over four decades in the treatment of toxicity from AAD and cocaine, hypertension and endocrine emergencies, CHF, stroke, and ACS. Given its ?1-, ?2-, and ?1-blocker properties, IV route of administration, widespread accepted use, and safety profile, labetalol represents a logical choice [155,161,166-172,176-180,183]. Labetalol is liphophilic and a Class II antiarrhythmic, with attenuation of sympathetic effects on the sinoatrial and atrioventricular nodes, as well as increasing non-pacemaker action potential duration and refractory period. Calcium channel blockers, phentolamine, nitroprusside, and nitroglycerin mitigate hypertension, but not uniformly tachycardia, for both AAD and cocaine, but the total number of studies is small.
The future should bring new understanding of AAD and adrenergic physiology, with resolution of the theoretical risk of ?unopposed ?-stimulation? after ?-blocker use in hyperadrenergic states. Prospective studies of the treatment of cardiovascular and CNS toxicity from AAD with ?- and ?-adrenoceptor blockers, calcium channel blockers, and various sedatives including dexmedetomidine, are critically needed to guide therapeutic recommendations with the goal of increasing patient safety and reducing length of stay.
Acknowledgement
The authors acknowledge the assistance of Judd E. Hollander, MD, University of Pennsylvania, for review and input on portions of this publication, and Robert S. Hoffman, MD, New York University, Rama B. Rao, MD, Weill Cornell Medical College, and Edward W. Boyer, MD, PhD, University of Massachusetts for their interest and expertise in prior communications regarding this topic.
References
1) http://www.unodc.org/documents/data-and-analysis/WDR2012/WDR_2012_web_small.pdf.
4) http://www.samhsa.gov/data/2k13/DAWN2k11ED/DAWN2k11ED.html
44) Matoba R (2001) [Cardiac lesions in methamphetamine abusers]. Nihon Hoigaku Zasshi 55: 321-330.
54) Bostwick DG (1981) Amphetamine induced cerebral vasculitis. Hum Pathol 12: 1031?1033.
127) (2004) Position paper: whole bowel irrigation. J Toxicol Clin Toxicol 42: 843-854.
207) Hollander JE (2008) Cocaine intoxication and hypertension. Ann Emerg Med 51: S18-20.