Research Article
Margarita Gutova1*, Davit Shahmanyan1, Diana Oganesyan1, Yelena Abramyants1, Lusine Danielyan2, William H. Frey II3, Vazgen Khankaldyyan4, Joseph Najbauer1, Irina V. Balyasnikova5, Rex A. Moats4, Maciej S. Lesniak5, Michael E. Barish1, and Karen S. Aboody1*
1Department of Neurosciences, Beckman Research Institute of City of Hope, Duarte, California, 91010, USA
2Department of Clinical Pharmacology, University Hospital of Tübingen, D-72076 Tübingen, Germany
3Neurosciences Research, HealthPartners Institute for Education and Research, St. Paul, Minnesota 55101, USA
4Department of Radiology, Children’s Hospital of Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, California 90027, USA
5Brain Tumor Center, University of Chicago, Chicago, Illinois 60637, USA
Corresponding author
Margarita Gutova, MD, Department of Neurosciences, Beckman Research Institute of City of Hope 1500 E. Duarte Rd., Duarte, CA 91010, USA, Tel: (626)-3598111 x65637; Fax: 626-471-7371; E-mail: mgutova@coh.org
Karen S. Aboody, MD, Department of Immunology and Biotechnology, University of Pécs Medical School, Pécs, Hungary, Tel: (626) 471-7177; Fax: (626) 301-8857; E-mail: kaboody@coh.org
Received Date: 13th February 2015
Accepted Date: 09th April 2015
Published Date: 15th April 2015
Citation
Gutova M, Shahmanyan B, Oganesyan D, Abramyants Y, Danielyan L, et al. (2015) Intranasal Delivery of Therapeutic Neural Stem Cells to Target Intracerebral Glioma. Enliven: J Stem Cells Regen Med 2(4): 006.
Copyright
@ 2015 Dr. Margarita Gutova. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Despite aggressive multimodal therapy and advances in imaging, surgical and radiation techniques, high-grade gliomas remain incurable, with patient
survival often measured in months. Treatment failure is largely attributable to the invasive nature of glioma cells, ineffective delivery of chemotherapeutic
agents across the blood-brain barrier, and dose-limiting systemic toxicities. Neural stem cells (NSCs) have inherent tumor-tropic properties that can be
exploited for targeted delivery of anti-cancer agents. However, current intracranial and intravenous injection approaches for administering NSCs are
not optimal, especially for repeat administrations, because the methods are invasive and may lead to complications. We hypothesized that intranasal
administration of NSCs would circumvent these challenges. In this study, we evaluated the biodistribution of NSCs administered intranasally to
severely immunodeficient esterase-deficient Esle mice bearing orthotopic xenografts of U251.eGFP.ffluc human gliomas. Histological imaging and
3D reconstruction revealed that NSCs specifically localized to tumor sites, but not to non-tumor areas of the brain. Importantly, mice treated with
intranasally administered NSCs that were genetically modified to express carboxylesterase, a prodrug-activating enzyme, in combination with CPT-11
showed increased survival and reduced tumor growth. These results support further development of intranasal delivery of NSCs as a new approach to
treating glioma and possibly other invasive brain tumors.
Keywords
Glioma; Brain tumor; Neural stem cells; Intranasal delivery; Targeted therapy; Enzyme/prodrug; Carboxylesterase; Irinotecan; CPT-11
Introduction
Stem cell therapies have shown much promise in preclinical animal models for the treatment of central nervous system (CNS) diseases and brain tumors [1]. The major challenge in developing such therapies has been effective delivery of therapeutic agents and cells to injury sites within the brain [2-4]. Many studies have shown that neural stem cells (NSCs) that are intracranially injected into the brain localize specifically to areas of CNS pathology [5-8]. However, intracranial injection is invasive and impractical for repeated dosing. Although intravenously administered NSCs have the potential to cross the blood-brain barrier (BBB) and localize to areas of damaged brain tissue, including tumors, this approach may lead to off-target systemic effects, as well as immune reactions toward allogeneic NSCs. Thus, although encouraging results have been obtained, current methods of NSC administration are imperfect and there is an urgent need for new methods of stem cell delivery. Intranasal administration has already been used to deliver proteins, growth factors, hormones and small molecules to the brain to treat neurological disorders [9] and has been adapted for the specific use of intranasal insulin to treat Alzheimer's disease [10]. In addition, intranasally administered mesenchymal stem cells (MSCs) can bypass the BBB to target the CNS, and their migration to CNS sites is enhanced by pretreatment with intranasal hyaluronidase [11]. Several research groups have used intranasal delivery of MSCs to successfully target and treat CNS damage in animal models of neonatal ischemia [12], Parkinson’s disease [13], ischemic stroke [14] and neonatal brain damage [15,16]. A recent study in an orthotopic mouse glioma model showed that intranasally administered MSCs that express TNF-related apoptosis-inducing ligand rapidly reach tumor sites and induce a cytotoxic effect in vivo [17]. Similarly, intranasally administered neural stem/progenitor cells have been reported to target intracerebral malignant gliomas in mice [18,19].
In the current study, we examined the migration of an established therapeutic clonal human NSC line (HB1.F3.CD) to human glioma tumors implanted into the frontal lobe of immunodeficient mice after intranasal administration. This HB1.F3.CD NSC line demonstrated safety in a first-inhuman safety/feasibility study when used in combination with 5-flurocytosine (5-FC) in patients with recurrent glioblastoma (clinical trial ID # NCT01172964). In the studies reported here, this same NSC line was further modified to express a modified human carboxylesterase (hCE1m6), which converts the CPT-11 (irinotecan) prodrug to the potent topoisomerase I inhibitor, SN-38 [20]. Following intranasal administration, these CE-expressing NSCs (NSC.CE) migrated specifically to tumor sites within the brain in two different immunodeficient mouse models and induced a cytotoxic effect when given in combination with CPT-11 in plasma esterase-deficient (Es1e) SCID mice. These results indicate that NSCs can be intranasally administered and used to target anti-cancer agents to intracranial tumors. Overall, intranasal delivery of NSCs may offer a more efficient and less invasive method of implementing stem cell therapies in many CNS diseases, which could significantly improve and expand stem cell-based therapeutic applications.
Materials and Methods
Neural Stem Cell Culture
HB1.F3.CD NSCs derived from a clinical equivalent research lot (City of Hope cGMP Facility) were thawed and cultured in T-175 tissue culture flasks in DMEM (Invitrogen, 10313-021; Carlsbad, CA), supplemented with 10% heat-inactivated FBS (Gemini, GBP-100500, West Sacramento, CA) and 2 mM L-glutamine (Invitrogen, 25030-081), and incubated at 37oC, 6% CO2 for 72 h prior to labeling with superparamagnetic iron oxide (SPIO) nanoparticles (Molday ION Rhodamine BTM; CL-50Q02-6A-50; BioPhysics Assay Laboratory, Inc., Worcester, MA). Control unlabeled HB1.F3.CD cells were cultured under the same conditions as the labeled cells. HB1.F3.CD cells (passage 25) were labeled with Molday ION as described previously (Cromer Berman SM et al. [21]). For therapeutic efficacy studies, HB1.F3.CD cells were adenovirally transduced to express a humanized form of carboxylesterase (hCE1m6) (Wierdl M et al. [20]).
Animal Models of Glioma Tumors
Beige nude XID and Es1e esterase deficient SCID mice were used to investigate the effect of various immunodeficient hosts on NSC migration. The severely immunodeficient beige nude XID strain was ideal for this study because it allowed us to test the stand-alone viability of intranasally delivered NSCs with minimal interference from the immune system. The beige nude XID strain lacks key immunological components such as B cells, macrophages, and lymphokine activated killer (LAK) cells. For therapeutic efficacy studies we used the less immunodeficient Es1e SCID mice, which lack B cells and T cells, similar to the beige nude XID strain, but possess NK cells, LAK cells, and macrophages.
Biodistribution Studies
To establish glioma tumor xenografts, U251.eGFP.ffluc human glioma cells (1 x 105 cells/2 μl) were implanted stereotactically (2 mm right of bregma, 1 mm rostral caudal and 2.5-3 mm deep) into the frontal lobe of mice. Fourteen days after tumor cell implantation, NSCs labeled with Molday ION Rhodamine BTM were administered intranasally (6 x 105 cells) in a total volume of 12 μl PBS (n= 8 mice). Two immunodeficient mouse hosts were used: beige nude XID mice and Es1e esterase deficient SCID mice (n=4 for each model). Intranasal administration of NSC.CEs was performed as previously described [21,22]. Briefly, NSCs (5 x 104 cells/1 μl) were applied to each nostril in doses of 3 μl with 2 min intervals after hyaluronidase treatment (100 units/mouse) (Sigma-Aldrich; H3757). Four mice were euthanized 24 h post-NSC administration, and 4 mice were euthanized 4 days after NSC administration. Brains were fresh frozen or embedded in paraffin. Serial 10-μm horizontal sections were made from the brains and processed for H&E staining, Prussian blue staining, and fluorescent imaging. Prussian blue staining was performed using the Accustain iron stain kit (Sigma-Aldrich, St. Louis, MO; HT20-1KT) according to the manufacturer’s protocol. Mouse peripheral organs (lungs, liver and spleen) and spinal cords were also collected, tissue was digested using proteinase K, and DNA was extracted and amplified by PCR using primers for v-myc to test for the presence of NSCs in non-tumor organs. The brain of a mouse injected intranasally with NSCs that were not labeled with Molday ION Rhodamine BTM was used for PCR analysis. Because the left hemisphere was not implanted with tumor, it was used as a control for the right frontal lobe, which had the implanted tumor. The GAPDH gene was used as a DNA loading control. DNA purified from HB1.F3.CD cells was used as a positive control for PCR amplification of the v-myc gene (replicon size 170 bp). Pure water was used as a negative control. PCR products were analyzed by agarose gel electrophoresis and staining of DNA bands with ethidium bromide. Therapeutic efficacy studies were performed using the same U251.eGFP.ffluc glioma xenograft model in Es1e esterase deficient SCID mice. Briefly, on day 1 mice received intracranial injections of U251.eGFP.ffluc glioma cells (1.5 x 105 cells/2 μl). Tumor growth was monitored by Xenogen imaging. All mice developed tumors were then intranasally administered NSC.CE cells on days 7 and 14 after tumor cells implantation. A bolus intravenous injection of CPT-11 (37.5 mg/kg) was given 4 days after NSC.CE injection (i.e., days 11 and 18). After two rounds of NSC.CE/CPT-11 treatment, mice were followed for long-term survival. Animals were monitored daily, to perform euthanasia on any animals showing objective signs of dying. The signs include: any obvious prolonged illness including such signs as lethargy, decreased fecal production, bleeding, dehydration, difficulty breathing, central nervous system disturbances, or chronic diarrhea or constipation, weight loss (> 20% body weight); scruffy hair coat, hunched posture, hypo or hyperthermia, impaired ambulatory movement, inability to remain upright. Animal meeting any of these criteria were euthanized, and the date was recorded as their death.
3D Reconstruction of Tumors and Brain Tissue
Three-dimensional (3D) reconstruction was performed using Reconstruct (version 1.1.0.1) software (Dr. Kristen M. Harris et al., SynapseWeb, Laboratory of Synapse Structure and Function, Austin, TX, USA; http://synapses.clm.utexas.edu). For each tumor, 7-11 serial brain sections (10 μm each) separated by 200 μm were imported into the Reconstruct program and aligned manually. Tumor alignment and NSC quantification were done on merged fluorescent image slides; auto-tracing of the tumor was done on eGFP and red-blue merged slides. NSC cells were identifies as red (Molday ION Rhodamine BTM –labeled), and DAPI staining was performed to visualize mouse brain tissue. To produce a 3D image, structures of interest were separated based on color (Prussian blue label for NSCs) and cell density (eGFP, tumor areas). Volumes containing these cells and structures were determined using Reconstruct.
Results
NSCs Migrate to Sites of Intracranial Glioma in Beige Nude XID Mice
All studies used the established NSC line HB1.F3.CD, which is a v-myc-immortalized, human clonal NSC line that has been genetically modified to stably express E. coli cytosine deaminase (CD) [23]. We have extensively characterized these NSCs as being genetically and functionally stable, non-tumorigenic, and minimally immunogenic [24]. The HB1.F3.CD line is established as a master cell bank at City of Hope, which should accelerate translation of promising findings toward clinical use.
First, we sought to determine if intranasally administered NSCs maintained their inherent tumortropism in vivo. HB1.F3.CD cells were iron-labeled with Molday ION Rhodamine B or feraheme to allow for detection by both Prussian blue staining and fluorescent visualization of NSCs in histological sections [25]. U251.eGFP.ffluc human glioma cells (1x105) were injected into the frontal lobe of beige nude XID mice using stereotactic coordinates as previously described (n=4) [26]. Fourteen days after tumor implantation, HB1.F3.CD cells were administered intranasally. Mice were euthanized one or four days after NSC administration. NSCs migrated to and distributed specifically at the tumor sites, but were not detected in non-tumor areas of the brain (Figure 1A - 1E). The presence of NSCs at the tumor site was determined by histological analysis of the brain sections. Two brains from the mice euthanized on day 1, were cryosectioned and stained with DAPI to visualize cell nuclei (Figure 1A). eGFP fluorescence was used to visualize the eGFP-expressing U251 tumor cells (Figure 1B). Molday ION Rhodamine B-labeled NSCs were detected (red) at tumor sites after NSCs injection by fluorescence imaging (Figure 1C), merged image (Figure 1D) and 3-dimensional (3D) reconstruction of serial histological sections for each brain were performed (Figure 1E). The brain of one mouse euthanized on day 4 post-NSC injection was analyzed by PCR to detect v-myc transcripts at the tumor sites; v-myc is a specific marker for the HB1.F3.CD cells (Figure 1F). PCR analysis using v-myc specific primers (replicon size 170 bp) showed that v-myc DNA was present in the right hemisphere, where tumor cells were implanted. However, v-myc was not detected in the left hemisphere, where tumor cells were not present (Figure 1F). Quantitative results showing the numbers of NSCs at the tumor site for each mouse are summarized in (Table 1).
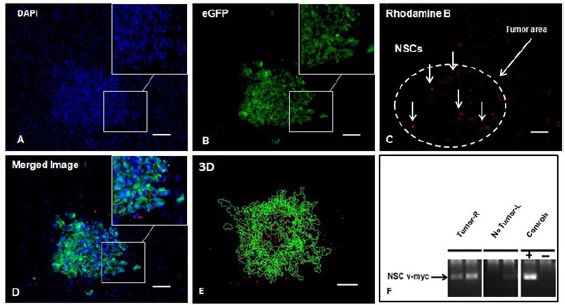
Severely immunodeficient beige nude XID mice (4 weeks old) were implanted intracranially with U251.eGFP.ffLuc human glioma cells. After 14 days, NSCs (6 x 105) labeled with Molday ION Rhodamine B were administered intranasally. Biodistribution of the NSCs was examined 24 h after administration and on day 4. Axial view of brain sections from the frontal lobe and containing tumor tissue (A) stained with DAPI (blue), (B) imaged for eGFP (green) to identify tumor cells, (C) imaged for Molday ION Rhodamine B (red) to identify NSCs. Vertical arrows indicate NSCs that migrated to the tumor site. Dashed white line outlines the tumor area. (D) Merged image. (E) Three-dimensional (3D) reconstruction of tumor and NSCs. (F) Nested PCR analysis for the v-myc gene (replicon size 170 bp) to determine the presence of HB1.F3.CD cells at the tumor site (right frontal lobe). No Tumor: left frontal lobe (no tumor); Controls: DNA purified from HB1.F3.CD cells (+) or no DNA added (-). Scale bars: 100 μm.
|
|
I |
II |
III |
IV |
|
|
Object |
Number of NSCs |
Number of brain sections |
Brain sections per tumor |
Total NSCs per brain |
Mice |
|
M01 day 1 |
170 |
9 |
180 |
3400 |
Beige nude XID |
|
M04 day 1 |
116 |
8 |
160 |
2320 |
Beige nude XID |
|
M07 day 1 |
209 |
11 |
220 |
4180 |
Es1e SCID |
|
M09 day 4 |
371 |
7 |
140 |
7420 |
Es1e SCID |
Migration of HB1.F3.CD Cells to Orthotopic Glioma Xenografts in Es1e SCID Mice
To determine if HB1.F3.CD cells also can migrate to glioma in mice capable of mounting a limited immune response or if they are rejected by residual immunological reactivity, we used Es1e SCID mice (n=4). Although these mice are immunodeficient, they have some ability to launch an immune response [27]. As early as 24 h after intranasal injection, HB1.F3.CD cells were identified in and around U251.eGFP.ffluc tumors in this model (Figure 2A). NSCs were not detected in the opposite hemisphere of the brain, which had not been implanted with tumor cells. Analysis of NSC migration four days after NSC administration revealed increased NSC accumulation at the tumor site (Figure 2C), when compared to brains of mice euthanized one day after injection of NSCs (Figure 2A and Table 1).
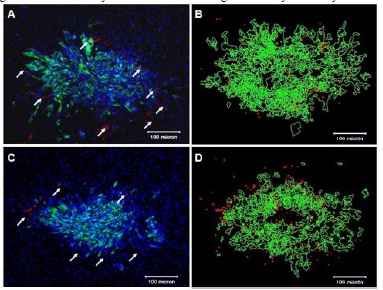
(A, C) SCID mice (Es1e) were implanted intracranially with U251.eGFP.ffluc cells (1x105 cells). Fifteen days after implantation of U251.eGFP. ffluc cells, NSCs (Molday-labeled HB1.F3.CDs, 6x105) were administered intranasally. Biodistribution of the NSCs was evaluated one (A) or four days (C) after NSC injection. White arrows in A and C indicate NSCs (red), tumor green and mouse brain blue (DAPI labeled). Scale bars: 100 μm. 3D tumor reconstruction of brain and NSCs that migrated to the tumor site one (B) or four days (D) after intranasal administration of NSCs.
We then used 3D reconstruction of fluorescent images of axial (horizontal) brain sections to quantitatively analyze the number of NSCs at the tumor sites (Figure 2B, 2D). Using images obtained from H&E and Prussian blue staining, we determined the brain area where the tumo was present. Brain sections (every 10th section) were selected for DAPI staining, fluorescen imaging and 3D analysis (Figure 2B, 2D). Fluorescent images (7-10 brain sections/mouse) were used for 3D analysis. Imaging was performed using blue, green and red channels to detect DAPI stained nuclei, tumor cells (U251.eGFP.ffluc green) or NSCs (Molday ION Rhodamine-labeled, red), respectively. Tumor alignment and quantification were done on DAPI-stained slides; auto tracing of the tumor was done on images using the green channel, and tracing of NSCs on redblue merged sections (Figure 2B and 2D; tumor pseudo-colored green and NSCs red). The number of NSCs was quantified in each brain section, and these values were multiplied by the number of sections. The total number of NSCs per brain was approximated based on the number of unstained intervening sections (Table 1). The same approach was used to estimate the numbers of NSCs present at the tumor sites in each mouse.
Detection of HB1.F3.CD NSCs in Peripheral Organs by Nested PCR
Nested PCR was used to detect the v-myc gene sequence (a marker for HB1.F3.CD NSCs) in the organs of mice that received intranasal administered NSCs (n=8). This approach could detect vmyc in 0.01 ng of genomic DNA purified from HB1.F3.CD NSCs. In our previous studies, we have estimated that nested PCR analysis of v-myc is sensitive enough to detect 10-20 NSCs per 500 ng of mouse brain tissue DNA [28]. Nested PCR failed to detect NSCs in the spinal cord as well, which was not surprising because no abnormalities were detected in this tissue by pathologic analysis. This approach did detect NSCs in tumor areas. NSCs were occasionally detected in the lungs of mice (e.g., a single lung in two mice [1/4 and 1/4 duplicates were positive], and may have been associated with pulmonary passage of intranasally administered NSCs on days 1 and 4 after NSCs injection (Supplementary Table 1, number of dashes indicate the duplicate samples). All PCR reactions were repeated 3 times (n=8). Further, PCR analysis of blood samples did not detect v-myc DNA in samples obtained 24-48 h after intranasal NSC administration, suggesting that NSCs were cleared from the bloodstream (data not shown). Therefore, we concluded that NSC distribution in the brain was specific to the U215 tumor areas after intranasal administration.
|
Sample ID |
M01 |
M03 |
M04 |
M05 |
M06 |
M07 |
M08 |
M09 |
|
Liver |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Lung #1 |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Lung #2 |
+ - - - |
+ - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Spleen |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Kidney #1 |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Kidney #2 |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
|
Spinal Cord |
- - - - |
- - - - |
- - |
- - |
- - |
- - |
- - |
- - |
Intranasal Administration of NSC.CEs in Combination with CPT-11 in Mouse Models of Glioma
We performed long-term survival studies using the U251 xenograft model to determine the effect of combination therapy with intranasally administered NSC.CE cells and CPT-11. NSC.CE cells (5x105) were administered intranasally to U251 tumor-bearing mice (n=8) on day 7 after tumor implantation, and the first dose of CPT-11 (37.5 mg/kg) was given on day 11 of the study as a bolus dose. The treatment with both NSC.CE cells and CPT-11 was repeated once the following week. Control groups included: 1) U251 tumor-bearing, no treatment (n=4); and 2) U251 tumorbearing, treatment with CPT-11 only (CPT-11 treatment was given on the same schedule as for mice that received NSC.CE + CPT-11) (n=8) (Figure 3). Statistical analysis of long-term survival data (using Log-rank Mantel-Cox test) showed that survival curves were significantly different for untreated vs. treated groups (p=0.02). However, median survival of mice treated with CPT-11 only and NSC.CE + CPT-11 was not statistically significant (median survival 47.5 days); median survival for the untreated group was 42.5 days. Further studies will be needed to optimize the treatment regimen for NSC-mediated CE/CPT-11 therapy.
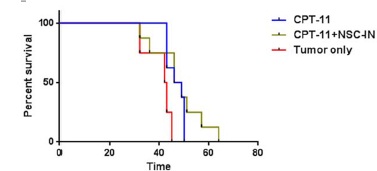
Figure 3. Long-term survival study of tumor-bearing mice treated with CPT-11 or NSC+CPT-11)
We performed long-term survival studies using the U251 xenograft model to determine the effect of combination therapy with intranasally administered NSC.CE cells and CPT-11. NSC.CE cells (5x105) were administered intranasally to U251 tumor-bearing mice (n=8) on day 7 after tumor implantation, and the first dose of CPT-11 (37.5 mg/kg) was given on day 11 of the study as a bolus dose. The treatment with both NSC.CE cells and CPT-11 was repeated once the following week. Control groups included: 1) U251 tumor-bearing, no treatment (n=4); and 2) U251 tumorbearing, treatment with CPT-11 only (CPT-11 treatment was given on the same schedule as for mice that received NSC.CE + CPT-11) (n=8) (Figure 3). Statistical analysis of long-term survival data (using Log-rank Mantel-Cox test) showed that survival curves were significantly different for untreated vs. treated groups (p=0.02). However, median survival of mice treated with CPT-11 only and NSC.CE + CPT-11 was not statistically significant (median survival 47.5 days); median survival for the untreated group was 42.5 days. Further studies will be needed to optimize the treatment regimen for NSC-mediated CE/CPT-11 therapy.
Discussion
In this study we provide evidence that, similar to biomolecules and MSCs, NSCs can use the intranasal pathway to gain access to the brain and to target brain tumors. Specifically, we have shown by fluorescent imaging, as well as PCR analysis, that after intranasal administration NSCs can migrate to gliomas implanted into the frontal lobe of mice. We have also demonstrated the potential for therapeutic efficacy of NSC.CEs administered intranasally to target U251 glioma tumors in mice. Intranasal administration of therapeutic NSCs provides several advantages over conventional methods, including intravenous and intracranial injections [29-31], and overcomes the inherent weaknesses of these methods. For example, intracranial administration involves invasive surgery of the brain and a significant proportion of stem cells are lost at the site of injection during surgery, resulting in an overall low rate of engraftment and survival of administered stem cells. Intranasal delivery of NSCs may also be superior to intravenous delivery methods because the host immune system is not exposed to the NSCs. The brain is considered an immunologically privileged organ, the CNS immune responses are generally primed in the periphery, and the access of circulating immune cells to the CNS is tightly controlled by the BBB [32]. Thus intranasal administration could minimize the likelihood of an immune response to such therapeutic cells. Further appeal of the intranasal method is that it allows NSCs to travel to the brain without triggering immunological responses in the rest of the body, and allows NSCs to bypass the challenge of crossing the BBB. Thus, current drawbacks to intracranial and intravenous delivery of NSCs may be partially overcome by using intranasal delivery. The HB1.F3.CD NSC line has been characterized for genetic stability, cellular identity, therapeutic gene expression, and tumor targeting ability [33-35]. We have also determined that HB1.F3.CD NSCs express HLA class I antigens but do not express detectable levels of class II antigens, indicating that they are unlikely to elicit a CD4+ helper T or CD8+ cytotoxic T cell response [34]. We have confirmed the feasibility of using intranasal delivery of therapeutic NSCs to target brain tumors. Prussian blue staining of iron nanoparticle-labeled NSCs, Molday ION Rhodamine B fluorescence imaging and PCR analysis confirmed that intranasally administered NSCs targeted sites of glioma (Figures 1 and 2). Further, we have provided evidence that after intranasal administration, NSCs migrate specifically to sites of glioma in the brain and do not appear to be present in non-tumor areas of the brain or other organs. Our data also suggest that intranasal delivery can potentially serve as a practical method of delivering therapeutic NSCs to the brain. Of note, the extended survival of glioma-bearing mice treated with intranasally administered NSC.CE cells in combination with CPT-11 as compared to control groups provides a foundation for further optimizing this novel therapy in pre-clinical models. Testing the intranasal NSC.CE + CPT-11 therapy in combination with other therapies to achieve a synergistic therapeutic effect is also warranted. We are further refining the intranasal delivery method, and are examining the clearance of NSCs through the circulation when they are injected intravenously. Intranasal delivery of NSCs represents a novel and promising tool for delivering NSCs to brain tumors for therapeutic approaches, such as enzyme/prodrug therapy of brain tumors [34]. If developed further, the intranasal delivery strategy could likely eventually be used to treat glioma patients in a minimally invasive and highly effective manner.
Acknowledgments
The authors acknowledge the technical expertise and advice of Sofia Loera of the Pathology Core supported by the National Cancer Institute of the National Institutes of Health award (P30CA33572) and the editorial assistance of Dr. Keely L. Walker (City of Hope). This work was supported by funding from the California Institute of Regenerative Medicine (DR1-01421), NIH/National Institute of Neurological Disorders and Stroke (U01 NS069997), Alex’s Lemonade Stand Foundation and Pediatric Cancer Research Foundation, Altschul Foundation.
Disclosure of Potential Conflicts of Interest
K.S.A. is a director and chief scientific officer of Thera Biologics, Inc., a clinical stage biopharmaceutical company focused on the development of stem cell-mediated cancer therapies.
References
- Aboody K, Capela A, Niazi N, Stern JH, Temple S (2011) Translating stem cell studies to the clinic for CNS repair: current state of the art and the need for a Rosetta Stone. Neuron 70: 597-613.
- Miller RH, Bai L (2012) Translating stem cell therapies to the clinic. Neurosci Lett 519: 87-92.
- Ming GL, Brüstle O, Muotri A, Studer L, Wernig M, et al. (2011) Cellular reprogramming: recent advances in modeling neurological diseases. J Neurosci 31: 16070-16075.
- Noble M, Davies JE, Mayer-Pröschel M, Pröschel C, Davies SJ (2011) Precursor cell biology and the development of astrocyte transplantation therapies: lessons from spinal cord injury. Neurotherapeutics 8: 677-693.
- Kim SU (2004) Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology 24: 159-171.
- Daniela F, Vescovi AL, Bottai D (2007) The stem cells as a potential treatment for neurodegeneration. Methods Mol Biol 399: 199-213.
- Vescovi A, Gritti A, Cossu G, Galli R (2002) Neural stem cells: plasticity and their transdifferentiation potential. Cells Tissues Organs 171: 64-76.
- Hawro T, Saluja R, Weller K, Altrichter S, Metz M, et al. (2014) Interleukin-31 does not induce immediate itch in atopic dermatitis patients and healthy controls after skin challenge. Allergy 69: 113-117.
- De Paepe ME, Luks FI (2013) What-and why-the pathologist should know about twin-to-twin transfusion syndrome. Pediatr Dev Pathol 16: 237-251.
- Schiöth HB, Craft S, Brooks SJ, Frey WH 2nd, Benedict C (2012) Brain insulin signaling and Alzheimer's disease: current evidence and future directions. Mol Neurobiol 46: 4-10.
- Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, et al. (2009) Intranasal delivery of cells to the brain. Eur J Cell Biol 88: 315-324.
- Liu CH, Chang CH, Kuo HC, Ro LS, Liou CW, et al. (2012) Prognosis of symptomatic patients with the A3243G mutation of mitochondrial DNA. J Formos Med Assoc 111: 489-494.
- Danielyan L, Schäfer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, et al. (2011) Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res 14: 3-16.
- Wu H, Li J, Zhang Q, Yan X, Guo L, et al. (2012) A novel small Odorranalectin-bearing cubosomes: preparation, brain delivery and pharmacodynamic study on amyloid-beta ?????-treated rats following intranasal administration. Eur J Pharm Biopharm 80: 368-378.
- Donega V, van Velthoven CT, Nijboer CH, Kavelaars A, Heijnen CJ (2013) The endogenous regenerative capacity of the damaged newborn brain: boosting neurogenesis with mesenchymal stem cell treatment. J Cereb Blood Flow Metab 33: 625-634.
- Donega V, Nijboer CH, van Tilborg G, Dijkhuizen RM, Kavelaars A, et al. (2014) Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Exp Neurol 261: 53-64.
- Balyasnikova IV, Prasol MS, Ferguson SD, Han Y, Ahmed AU, et al. (2014) Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol Ther 22: 140-148.
- Reitz M, Demestre M, Sedlacik J, Meissner H, Fiehler J, et al. (2012) Intranasal delivery of neural stem/progenitor cells: a noninvasive passage to target intracerebral glioma. Stem Cells Transl Med 1: 866-873.
- Cova L, Bossolasco P, Armentero MT, Diana V, Zennaro E, et al. (2012) Neuroprotective effects of human mesenchymal stem cells on neural cultures exposed to 6-hydroxydopamine: implications for reparative therapy in Parkinson's disease. Apoptosis 17: 289-304.
- Wierdl M, Tsurkan L, Hyatt JL, Edwards CC, Hatfield MJ, et al. (2008) An improved human carboxylesterase for enzyme/prodrug therapy with CPT-11. Cancer Gene Ther 15: 183-192.
- Cromer Berman SM, Kshitiz, Wang CJ, Orukari I, Levchenko A, et al. (2012) Cell motility of neural stem cells is reduced after SPIO-labeling, which is mitigated after exocytosis. Magn Reson Med 69: 255-262.
- Aboody KS, Najbauer J, Metz MZ, D'Apuzzo M, Gutova M, et al. (2013) Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med 8: 184ra159.
- Campbell C, Morimoto BH, Nenciu D, Fox AW (2012) Drug development of intranasally delivered peptides. Ther Deliv 3: 557-568.
- Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, et al. (2011) Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther 19: 1769-1779.
- Shingaki T, Inoue D, Furubayashi T, Sakane T, Katsumi H, et al. (2010) Transnasal Delivery of Methotrexate to Brain Tumors in Rats: A New Strategy for Brain Tumor Chemotherapy. Mol Pharm 7: 1561-1568.
- Serwer LP, James CD (2012) Challenges in drug delivery to tumors of the central nervous system: an overview of pharmacological and surgical considerations. Adv Drug Deliv Rev 64: 590-597.
- Ransohoff RM, Engelhardt B (2012) The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12: 623-635.
- Aboody KS, Najbauer J, Danks MK (2008) Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther 15: 739-752.
- Gutova M, Shackleford GM, Khankaldyyan V, Herrmann KA, Shi XH, et al. (2013) Neural stem cell-mediated CE/CPT-11 enzyme/prodrug therapy in transgenic mouse model of intracerebellar medulloblastoma. Gene Ther 20: 143-150.