Research Article
Hiroki Miyashita1, Robert H. Kretsinger, MD2, Norio Matsushima1*
1 The Institute of Tandem Repeats, Sapporo 004-0882, Japan
2 Department of Biology, University of Virginia, Charlottesville 22904, USA
Corresponding author
Norio Matsushima, Biophysicist, The Institute of Tandem Repeats, Sapporo 060-8556, Japan, Tel: +81 11 886 0087; Fax: +81 11 886 0087; E-mail: norio_irreko@outlook.jp
Tel: 9322211472;
E-mail: drpravinubale@gmail.com;
Received Date: 07 October 2014
Accepted Date: 07 November 2014
Published Date: 10 November 2014
Citation
Miyashita H, Kretsinger RH, Matsushima N (2014) Comparative Structural Analysis of the Extracellular Regions of the Insulin and Epidermal Growth Factor Receptors whose L1 and L2 Domains have Non-Canonical, Leucinerich Repeats. Enliven: Bioinform 1(4): 005
Copyright
© 2015 Dr. Pravin Ubale. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Insulin receptor (IR) and epidermal growth factor receptors (EGFR) are members of the receptor tyrosine kinase super family. The extracellular regions of both IR and EGFR contain two L domains. Many crystal structures of the extracellular regions of the IR and EGFR families have been determined in both the unliganded state and in complexes with ligands. The structures reveal that the L domains consist of four to six leucine rich repeats (LRRs); although, their amino acid sequences are highly variable. The present bioinformatic analysis reveals some features on the LRRs and the structures. We conclude that the LRRs in the L domains belong to a non-canonical motif differing from the known (canonical) motifs; the repeat units consist of two β-strands and the overall shape of the LRRs resembles a prism. To characterize the spatial arrangement of the two L-domains we propose two parameters; the distance between the two L domains (D) and the angle between the two axes showing the direction of the β-sheet stacking of the LRRs in the L domains (Ψ). These two parameters, D and Ψ, describe an essential feature of the structures and ligand induced structural changes.
Keywords
Insulin receptor;?Epidermal growth factor receptor; L domain; Leucine-rich repeats; Non-canonical LRR; Ligand interaction; Dimer; Toll-like receptor; Geometric analysis
Abbreviations
CR: Cys-rich Region; EGF: Epidermal Growth Factor; EGFR: Epidermal Growth Factor Receptor; FnIII: Fibronectin type III domains; HCS: Highly Conserved Segment; IGF-1R: Insulin-like Growth Factor 1-Receptor; IR: Insulin Receptor; IRR: Insulin Receptor-related Receptor; LRR: Leucine-Rich Repeat; Nrg-1: Neuregulin-1; 3D: Three Dimensional; TGF? : Transforming Growth Factor-? :TLR: Toll-Like Receptor; VS: Variable Segment
Introduction
The insulin receptor (IR) and epidermal growth factor receptor (EGFR) families are both members of the receptor tyrosine kinase super family [1-6]. The IR is a large, transmembrane, glycoprotein dimer consisting of several structural domains (figure 1). The N-terminal half of the ecto-domain contains two L domains (L1 and L2) separated by a cys-rich region (CR). The C-terminal half of the IR ectodomain consists of three fibronectin type III domains (FnIII), the second of which contains an insert region of about 120 residues. The IR family consists of IR, the insulin like growth factor 1-receptor (IGF-1R) and the insulin receptor related receptor (IRR).?The IR and IGF-1R interact with insulin, IGF-I, and IGF-II. The IR mediates the pleiotropic actions of insulin. The IGFs act via IGF-1R to promote cell proliferation, survival, and differentiation. Binding of insulin to the IR leads to phosphorylation of several intracellular substrates, including, insulin receptor substrates (IRS1-4), SHC, GAB1, CBL and other intermediates involved in cell signaling. The IR family undergoes processing to form two polypeptide chains, α and β, that are assembled into a hetero-tetramer, or an (αβ)2 homodimer, stabilized by disulfide bonds (figure 1).
Figure 17
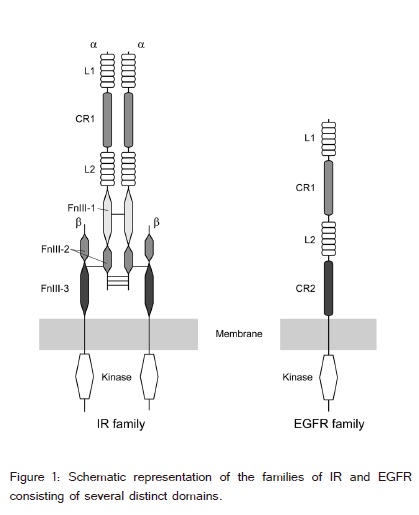
The EGFR ectodomain contains four domains - L1, CR1, L2 and CR2 (figure 1). The L1 and L2 domains are homologous [7]. The EGFR family consists of EGFR (ErbB1/ HER1), ErbB2 (HER2/Neu), ErbB3 (HER3) and ErbB4 (HER4), and plays important roles in cell growth, differentiation, survival, and migration [8,9].The ErbB receptors interact with eleven polypeptide growth factor ligands including epidermal growth factor (EGF), transforming growth factor-α(TGFα) and neuregulin-1 (Nrg-1) [5]. Drosophila ErbB2 binds an antagonist, Spitz [10]; although, ErbB2 lacks a known ligand [11]. The EGFR family is activated by ligand-induced dimerization of the receptors; however, there is increasing evidence that the EGFR family is present as pre-formed, inactive, dimers prior to ligand binding [1].
The IR and EGFR family have multiple, intra chain disulfide bonds and N-linked glycosylation sites. A large number of crystal structures of the extracellular regionsof eight proteins of the IRs and EGFR families have been determined in the unliganded state and in complexes with ligands[12-43].Moreover, their structures in complex with various (therapeutic) antibodies including Cetuximab and Herceptin have been determined [15,17,21-23,25,29,31,32,35-39,41-43]. The unliganded extracellular regions of EGFR, ErbB3, and ErbB4 all adopt a characteristic ?tethered? conformation in which the primary receptor dimerization site is occluded by intramolecular interactions between CR1 and CR2 [21]. Upon ligand binding, the receptor undergoes a dramatic rearrangement of domains. The EGFR ligands, including EGF, bind simultaneously to both L1 and L2, forcing them to adopt the extended configuration that is capable of CR1-mediated dimerization.
The structures reveal that these L domains consist of four to six LRRs; although, the LRRs show extreme variability with major insertions in some of their repeats.?LRRs are present in over 60,000 proteins. Each repeat of LRRs is typically 20-30 residues long and can be divided into an HCS (Highly Conserved Segment) and VS (Variable Segment). The HCS part consists of LxxLxLxxNx(x/-)L, in which ?L? is Leu, Ile, Val, or Phe, ?N? is Asn, Thr, Ser, or Cys, ?x? is a non-conserved residue, and ?-? is a possible deletion site [44-46]. There are eight classes of LRRs-?Typical?, ?SDS22-like?, ?IRREKO?, ?Bacterial?, ?Plant specific?, ?TpLRR?, ?RI-like?, and ?Cysteine-containing? [46-48]. Tandem LRR domains consist of a super helical arrangement of repeating structural units and fold into a horse shoe, a right-handed or left-handed helix, or a prism shape [49]. Three residues at positions 3 to 5, xLx, in the HCS part form a short β-strand. These β-strands stack parallel and then the LRRs assume their super-helical arrangements.
Very recently, Miyashita et al.[50] identified novel LRRs in over three hundred proteins from unicellular eukaryotes and bacteria. The HCS clearly differs from the canonical motifs with the consensus of VxGx(L/F)x(L/C)xxNx(x/-)L that is characterized by the addition of Gly between the first conserved Val and the second conserved Leu. However, the structure remains unknown.?
Cellular Automata (CA) is a basic model of a spatially developed decentralized system, made up of various unique components called Cells.
There have been many determinations of the 3D structures of the IR and EGFR family [12-43]. Moreover, excellent reviews have been prepared in the families of IR and/or EGFR receptors [1-6]. However, it appears that there is no geometric analysis to provide quantitative analysis and comparison of the 3D structures.
Here we show that LRRs in the L domains belong to the non-canonical motif and describe some features of this structure. We propose two parameters to characterize the spatial arrangement of the two L-domains. The two parameters provide fundamental features of the ligand interactions in the IR and EGFR families.
Methods
Sequence Alignment and Secondary Structure
There were forty-three PDB files for the ectodomains of the IR and EGFR families in the NCBI on June 24, 2014 (http://www.ncbi.nlm.nih.gov/). The files contain eight different proteins. They are IR and IGF-1R from human, human EGFR, ErbB2 from human, rat, and Drosophila, human ErbB3, and human ErbB4. The 3D structures of almost the entire ectodomains or the parts (such as L1-CR-L2 in the IR or L2 in the EGFR) have been determined [12-43].
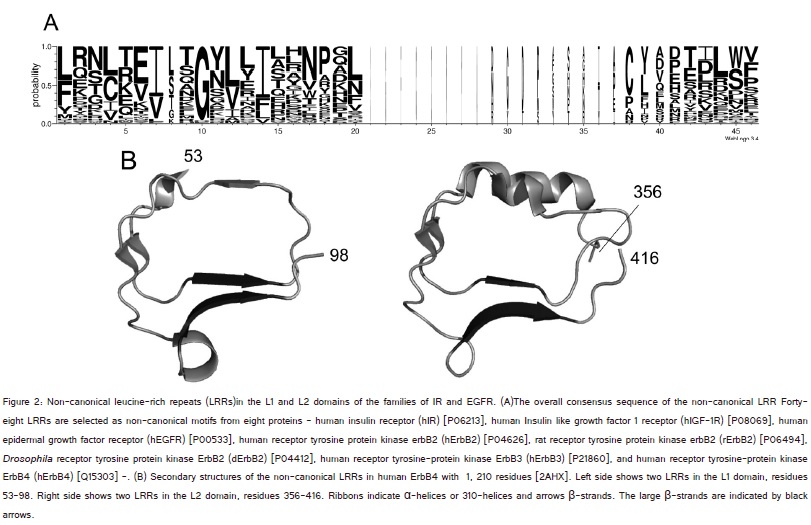
Secondary structure assignment from the atomic coordinates of the IR and EGFR proteins were made by DSSP (http://crdd.osdd.net/raghava/ccpdb/beta2_up.php) (figure 2) [51].?Sequence alignments of individual LRRs were made by MAFFT (http://www.genome.jp/tools/mafft/) (Supplementary material) [52] and finally by eye.
Figure 18
Geometric Analysis
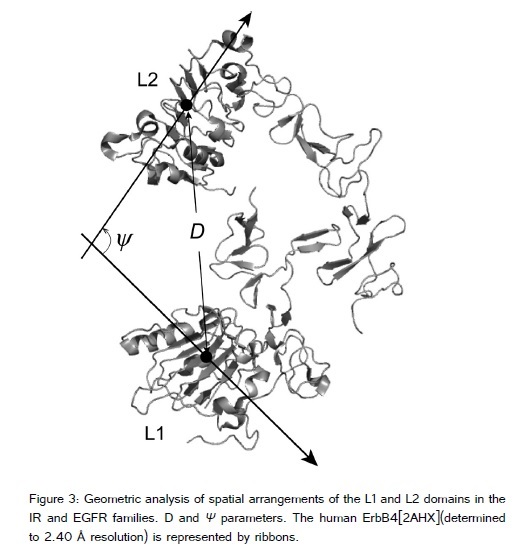
To understand the spatial arrangement of the L1 and L2 domains we propose two structural parameters, ?D? and ?ψ? (figure 3). ?D? is the distance between the L1 and L2 domains. ?ψ? is the angle between the two axes that indicate the direction of parallel stacking of the β-strands in the two L domains. The axis was calculated by a straight line fitting program in MATLAB (MathWorks) that fits a line in 3D space to a set of data points. The coordinates of the α-carbon (Cα) of the consensus leucine residue at position 5 (corresponding to the middle of each β-strand that forms a large β-sheet) in individual repeat units were used as the data points. ?D? is the distance between the center of gravity in the two L domains (as defined by the average Cαcoordinates of LRRs in individual L domains).
figure-19
Results and Discussion
Non-Canonical LRR in the L1 and L2 Domains
The sequence analysis predicted six potential LRRs in the respective L domain[16]. However, the structures reveal that in the IR families the L1 domain contain six LRRs and the L2 contains four or five LRRs, in which the β-strands of these LRRs stack parallel, while in the EGFR families L1 has four or five LRRs and L2 has five (table 1).The N- or C-terminal units of the six potential LRR unitsin the L domains usually do not form a β-strand and thus cannot participate in the stacking of parallel β- strands.
| Protein | Domains | Na | L1 b |
L2 c |
Dd (Å) |
ψe (º) |
Ligand/ Comments |
PDB | Resolution (Å) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Human IR | L1-CR1-L2 | 11 | 6 | 5 | 55.7 | 112 | Free state | 2HR7_A | 2.32 |
| ? | L1-CR1-L2 | 11 | 6 | 5 | 49.6 | 141 | ? | 2HR7_B | ? | |
| ? | L1-CR1-L2-(FnIII-1)-(FnIII-2) (achain) | 10 | 6 | 4 | 45.8 | 125 | Fab 83-7 heavy and light chains | 3LOH_E | 3.8 | |
| ? | L1-CR1-L2-(FnIII-1)-(FnIII-2) (a chain) | 11 | 6 | 5 | 43.6 | 125 | Fab 83-7 heavy and light chains | 2DTG | 3.8 | |
| ? | L1-CR1-L2-(FnIII-1) | 11 | 6 | 5 | 49.8 | 162 | Insulin A and B chains, Fab 83-14- heavyand light chains | 3W14_E | 4.4 | |
| ? | L1 - CR1 - L2 - (FnIII-1) | 11 | 6 | 5 | 49.5 | 160 | ? | 3W14_F | ? | |
| 2 | Human IGF-1R |
L1 -CR1 - L2 | 11 | 6 | 5 | 51.1 | 103 | Free state | 1IGR_A | 2.6 |
| 3 | Human EGFR | L1 -CR1 - L2 - CR2 | 10 | 5 | 5 | 66.7 | 135 | Cetuximab Fab lightchain and Fab heavy chain | 1YY9 | 2.61 |
| ? | L1-CR1-L2-CR2 | 9 | 4 | 5 | 59.6 | 111 | ? | 4KRP_A | 2.82 | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 61 | 111 | Cetuximab heavy and light chains, Nanobody/VHH domain EgA1 | 4KRO_A | 3.05 | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 56.7 | 112 | Adnectin | 3QWQ_A | 2.75 | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 55.7 | 93 | EGF/ | 1NQL | 2.8 | |
| Inactive?(lowpH) complex | ||||||||||
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.7 | -140 | EGF | 3NJP_A | 3.3 | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.7 | -139 | " | 3NJP_B | " | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.6 | -140 | EGF | 1IVO_A | 3.3 | |
| ? | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.7 | -139 | " | 1IVO_B | " | |
| ? | L1-CR1-L2 | 9 | 4 | 5 | 30.6 | -144 | TGFa | 1MOX_A | 2.5 | |
| ? | L1-CR1-L2 | 9 | 4 | 5 | 29.9 | -138 | ? | 1MOX_B | " | |
| 4 | Human Erb-B2 |
L1-CR1- L2-CR2 | 10 | 5 | 5 | 26.7 | 109 | Herceptin Fab heavy and light chains, | 1N8Z_C | 2.52 |
| " | L1-CR1- L2-CR2 | 10 | 5 | 5 | 27.5 | 104 | Fab37 heavy and light chains | 3N85_A | 3.2 | |
| " | L1-CR1- L2-CR2 | 10 | 5 | 5 | 27.3 | 103 | Pertuzumab Fab heavy and light chains | 1S78_A | 3.25 | |
| " | L1-CR1- L2-CR2 | 10 | 5 | 5 | 27.5 | 103 | " | 1S78_B | " | |
| " | L1-CR1- L2-CR2 | 10 | 5 | 5 | 27.5 | 104 | Immunoglobulin G-binding protein A | 3MZW_A | 2.9 | |
| " | L1-CR1- L2-CR2 | 10 | 5 | 5 | 27.2 | 107 | Fab heavy and light chains | 3BE1_A | 2.9 | |
| " | L1 -CR1 - L2 | 10 | 5 | 5 | 26.9 | 106 | Free state | 2A91 | 2.5 | |
| 5 | Rat Erb-B2 | L1-CR1-L2-CR2 | 10 | 5 | 5 | 27.3 | 105 | Free state | 1N8Y_C | 2.4 |
| 6 | Drosophila Erb-B2 | L1-CR1-L2-CR2 | 10 | 5 | 5 | 31.6 | 120 | Free state | 3I2T_A | 2.7 |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 29.5 | -119 | Protein spitz | 3LTF_A | 3.2 | |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 29.7 | -126 | ? | 3LTF_C | " | |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 30 | -120 | Protein spitz | 3LTG_A | 3.4 | |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 31.1 | -127 | ? | 3LTG_C | " | |
| 7 | Human Erb-B3 |
L1-CR1-L2-CR2 | 10 | 5 | 5 | 69.3 | 148 | Free state | 1M6B_A | 2.6 |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 69.6 | 148 | " | 1M6B_B | " | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 69.1 | 157 | RG7116 Fab heavy and light chains | 4LEO_C | 2.64 | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 72.4 | 150 | Fab heavy and light chains | 4P59 | 3.4 | |
| " | L1-CR1-L2 | 10 | 5 | 5 | 69.8 | 133 | Fab DL11 heavy and light chains | 3P11_A | 3.7 | |
| 8 | Human Erb-B4 |
L1-CR1-L2-CR2 | 10 | 5 | 5 | 68.2 | 112 | Free state | 2AHX_A | 2.4 |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 70.1 | 113 | " | 2AHX_B | " | |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 68 | 121 | Fab heavy and light chains | 3U9U_E | 3.42 | |
| " | L1-CR1-L2-CR2 | 9 | 4 | 5 | 67.1 | 110 | " | 3U9U_F | " | |
| " | L1-CR1-L2 | 9 | 4 | 5 | 59 | 125 | Free state | 3U2P_A | 2.57 | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.7 | -127 | Nrg-1 | 3U7U_A | 3.03 | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.4 | -127 | " | 3U7U_B | " | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.6 | -127 | " | 3U7U_C | " | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.4 | -126 | " | 3U7U_D | " | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.7 | -130 | " | 3U7U_E | " | |
| " | L1-CR1-L2-CR2 | 10 | 5 | 5 | 32.5 | -127 | " | 3U7U_F | " |
The consensus sequence may be represented by IxGxLxIxxNxLxxxxxxxxxL/FxxL/Cxxand the segments of IxGxL and NxL are frequently replaced by IxxGxL and ?NxxL, respectively (figure 2A). Gly at position 3 is highly conserved. The VS is more variable. The HCS consists of a twelve residue stretch that is characterized by the addition of Gly between the first conserved hydrophobic Ile and the second conserved hydrophobic Leu. This is consistent with non-canonical motifs proposed by us [50]. Three residues at positions 4 to 6, xLx, in the HCS (corresponding tothree residues at positions 3 to 5 in the canonical HCS) form a β-strand that is part of a large β-sheet (figure 2B). Moreover, two residues, xI, in which ?x? is at the last position in the preceding repeat and ?I? is at position 1, form an additional β-strand (figure 2B).
Most of the repeat units including the non-canonical units contain both a short β-strand of two to three residues and a longer β-strand of three to five residues forming large β-sheet (figure 2B). This β-β structural motif clearly differs from those of the other (canonical) LRR classes [49].
The non-canonical LRRs in individual L domains have two β-sheets. One consists of the parallel stacking of β-strand at positions 4 to 6, xLx, in the HCS (called the large β-sheet). The other is parallel β-strand stacking of the two residues xI. The overall shape of the non-canonical LRR domains resembles a prism (figure 3), as seen in the ?TpLRR? class[49]. A prism is also formed by β-helices consisting of other types of tandem repeats in proteins. A β-helix is a protein structure formed by the association of parallel β-strands in a helical pattern with two, three, or four faces [51-54].
Structural Features of the IR and EGFR Families
The structural features in the unliganded state:
The crystal structures of the extracellular regions of the eight proteins of the IR and EGFR families are available in the unliganded state [12-14,16,17,22,23,26,27,29-33,35-40,43].The extracellular regionsof the IR family in the crystals consist of L1-CR1-L2, L1-CR1-L2-(FnIII-1), orL1-CR1-L2-(FnIII-1)-(FnIII-2) (? chain), while those of the EGFR family contain L1-CR1-L2 or L1-CR1-L2-CR2 (table 1). The structures of L1-CR1 (in IR), L2 (in EGFR), and L1 (in Erb-B2) have also been determined [15,21,22,25,31,41,42].
The D?s of the IR family are smaller than those of the EGFR family except for ErbB2. The D (= 27Å) of ErbB2 is the smallest among the known structures of the receptors in the unliganded state. In contrast, the IR family has largerψs?s than does the EGFR family. The unliganded ErbB2sadopt an extended conformation as do the complexes of EGFR with EGF or TGFα and of the complex of Erb-B4 with Ngr-1. Consequently, the D of the unliganded ErbB2 is comparable to those of the EGFR and ErbB4 complexes, whileψ is slightly smaller.
Structural changes induced by ligand-interactions
Structures of the human IR have been reported in complexes with insulin (figure 4A) [15]. Insulin interacts directly with the large β-sheet of the L1 domain. The αCT segment, residues 693-710, also contributes to interaction with insulin [13]. The structures of EGFR complexes bound to ligands - EGF, TGFα, Nrg-1, or Spitz - have been determined (figure4B) [18-20,24,28]. In the EGFR family, the ligands directly interact with both the L1 and L2 domains of two intramolecular LRR domains through side chains of the residues involved in the parallel stacking of the β-strands forming the large β-sheet. Two intermolecular LRR domains also contribute to interactions with ligands including dsRNA and myeloid differentiation protein 2 (MD-2) in toll-like receptors (TLRs), and consequently the TLRs form homo-, or hetero-dimers [55-61].
Figure 20:
The IR family is activated by ligand induced dimerization of the receptors [1-3]. The active dimeric form (in the liganded state) has almost equal or slightly smaller D than in the inactive dimeric form (in the unliganded state) and has largerψ(table 1 and figure 4A). The conformational change is small in comparison with that of the EGFR, as extensively reviewed by Ward et al. [6]. Limited conformational change in IR upon hormone binding is compatible with a small angle X-ray scattering study of IGF-I binding to the soluble IGF-IR ectodomain [62].Thorough studies indicate that members of the EGFR family are present as pre-formed, yet inactive, dimers prior to ligand binding, as reviewed by Maruyama [1]. He proposed two models for the mechanism of the activation the EGFR. In the models the active form is dimeric in the liganded state. The D?s of the active forms of human EGFR and Erb-4 are smaller than those of the inactive forms, while the ψ's?are larger (table 1 and figure 4B). The D and ψof the EGFR in an inactive (low pH) complex with EGF are more similar to those in the unliganded state than in the active liganded state. Furthermore, the D and ψof the liganded Drosophila Erb-2 are comparable with the unliganded form (table 1).
Consequently, the structural changes induced by protein, ligand interactions may be grouped into three categories based on just two parameters -?D = D (liganded) ? D (unliganded) and ?ψ= ψ(liganded) - ψ(unliganded) (table 1). The first category is seen in the IR-insulin complex where?D? 0Å and ?ψ ? 34°. The second category is seen in the complexes of EGFR with EGF or TGF? and of ErbB4 with Ngr-1; ?D ? -30Å and ?ψ ? 110°. The ErbB2-Spitz complex illustrates the third category; ?D ? 0 Å and ?ψ ? 0°.
(Therapeutic) antibody interactions
The structures of the EGFR family revealed that the antibodies interact with the L1 domain or the L2 domain in which the extracellular regions adopt the ?tethered? conformation, except for ErbB2. Consequently, the antibodies block the ligand binding site on the L1 or L2, and can prevent the ligands from interacting with the receptors. In addition, the antibodies will sterically prevent the extra-celluar region of the receptors from adopting the extended conformation that is required for dimerization [21].The values of the parameters ?D? and ?ψ?of ErbB3 and ErbB4 in complex with the antibodies are very comparable to those in the free state, as expected (table 1).
ErbB2 in complex with antibodies also adopts the extended conformation in the free state and in the liganded state. The two parameters do not change due to the formation of the complex. Thus, the antibodies do not interact with the L1 and the L2 domains.
Conclusions
The L1 and L2 domains of the IR and EGFR families consist of four to six LRRs. These LRRs have non-canonical motifs. Each repeating unit is represented by the structural unit β-βThe overall shape of the LRRs resembles a prism. The spatial arrangement of the two L-domains is well characterized by the two parameters consisting of the distance between the two L domains (D) and the angle between the two axes of the β-sheet stacking in the L domains (ψ).
Docking analysis revealed that incensole docks well with human aldose reductase (ALR2) and it interacts through hydrogen bonding. This interaction leads to the formation of stable ALR2-incensole complex. Thus it is a good molecule and it can be considered for developing into a potent human aldose reductase inhibitor to relief the diabetes long term complications.
References
47. Kajava AV (1998) Structural diversity of leucine-rich repeat proteins. J Mol Biol 277: 519-527.