Short Communication
Daniela Karall1*, Sabine Scholl-Bürgi1, Consolato Sergi3, Ralf Geiger2, Georg Engl2, Thomas Karall2, Joerg-Ingolf Stein2, Frederic M. Vaz4 and Ulrich Schweigmann2
1Medical University of Innsbruck, Clinic for Pediatrics I, Inherited Metabolic Disorders
2Medical University of Innsbruck, Clinic for Pediatrics III, Cardiology
3Medical University of Innsbruck, Institute of Pathology
4Academic Medical Center, Amsterdam, Lab Genetic Metabolic Diseases
Corresponding author
Daniela Karall MD, Clinic for Pediatrics I, Inherited Metabolic Disorders, Medical University of Innsbruck, Anichstrasse 35, A-6020 Innsbruck, Austria, Tel: +4351250423600; Fax: +4351250424941; E-mail: daniela.karall@i-med.ac.at; daniela.skladal@i-med.ac.at
Received Date: 31st October 2014
Accepted Date: 25th November 2014
Published Date: 30th November 2014
Citation
Karall D, Scholl-Bürgi S, Geiger R, Engl G, Karall T, et al. (2014) Barth Syndrome and Left-Ventricular Non-Compaction: Case Report and Surveillance Plan Prior to Cardiac Transplantation. Enliven: Surg 1(2): 004
Copyright
@ 2014 Dr. Daniela Karall. This is an Open Access article published and distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
Abstract
Barth syndrome, an X-linked disorder of cardiolipin metabolism, shows cardioskeletal cardiomyopathy, neutropenia, psychomotor and growth retardation. A limiting factor is heart function. We present a ten year old boy who underwent heart transplantation after bridging with an assist device at 8 months, and propose a surveillance plan prior to transplantation.
Keywords
Barth syndrome; Heart transplantation; Assist device; Left ventricular non-compaction
Introduction
Barth syndrome (BS, OMIM #302060), originally described in 1983, is a rare X-linked inherited disorder characterized by cardioskeletal myopathy, peripheral neutropenia, psychomotor and growth retardation and 3-methylglutaconic aciduria [1]. The number of cases is approximately 100 (personal communication from the Barth Syndrome foundation, cited by Yen [2]). The causative gene of this syndrome TAZ (G4.5) is located on Xq28 and its gene product taffazin is a homologue of the glycerolipid acyltransferases superfamily. It is involved in the phospholipid metabolism and in association with another gene LDB3 seems involved in the early heart development [3]. Barth syndrome is also characterized by the incapacity of mitochondria to produce adequate amounts of tetralinoleoyl-cardiolipin, an essential lipid for normal mitochondrial structure and energy production; linking the dilated cardiomyopathy in taffazin deficiency to the reactive oxygen species produced by dysfunctional mitochondria.
Here we report an additional case of Barth syndrome, which is unique, because we report the longest follow-up after successful bridging and cardiac transplantation in a patient with Barth syndrome and left-ventricular non-compaction (LVNC) on explanted heart. In addition, we also propose a surveillance plan prior to the heart transplantation in these patients.
Case Report
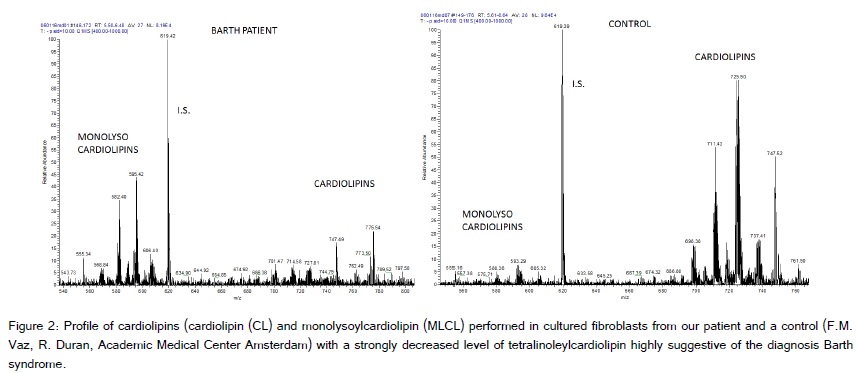
The patient is the first child of healthy non-consanguineous Caucasian parents, born after an uneventful pregnancy in the 40th gestational week through vaginal delivery with Apgar scores of 10/10/10, umbilical arterial pH 7.23 and birth weight of 3.270 g, length 51 cm, head circumference 33 cm. On the third day of life he was transferred to a tertiary care hospital due to grunting and tachypnea. Acid-base metabolism showed a pH of 7.15, base excess of -15 mmol/L and lactate of 148 mg/dL (15-22) (= 16.36 mmol/L). The body weight at admission was 3.045 g. Besides tachypnea and some apneic episodes there were no clinical symptoms. However, he needed slight oxygen supply (flow ½ liter/min), that increased when physically more active. No seizures were recorded. Acidosis was repeatedly corrected with sodium bicarbonate over the next three days and lactate normalized (15 mg/dL). Initial lumbar puncture and sepsis workup were normal. Blood chemistry showed normal values. Pyruvate was initially elevated [2.37 mg/dL (0.36-0-59) (= 262 µmol/L)]. Plasma, urine and CSF amino acids were basically normal (with some nonspecific minor changes). The analysis of urinary organic acids showed a marked lactaturia, no dicarboxylic aciduria or excretion of other metabolites, especially no methylglutaconic acid. In several repeat samples at the age of one, two, three and four months some methylglutaconic acid was found (not quantified). Results of newborn screening and otoacustic emissions were normal. Karyogram was 46, XY. ECG showed slight ST decrement and physiological signs of right ventricular hypertrophy. Transthoracic echocardiography revealed normal cardiac structure and function with closed ductus arteriosus with a minor pericardial effusion. Left ventricular non-compaction was not a prominent feature, even a few weeks before heart transplantation (Figure 1). On chest X-ray cardial transversal width was enlarged. There is no causal treatment for Barth syndrome or mitochondrial disorders in general, however, a supportive treatment with cofactors and vitamins of the respiratory chain is often introduced in these patients to possibly stabilize their mitochondrial function. Thus, our patient was given thiamine 1 x 100 mg, riboflavine 1 x 50 mg, vitamin C 1 x 200 mg, L-carnitine 2 x 150 mg and vitamine D3 1 x 800 IU. The child was dismissed fully breast-fed on day 18 of life with a weight of 3.770 g, length 53 cm (both values 50th percentile), head circumference 33.5 cm (10th percentile). The profile of cardiolipins performed in cultured fibroblasts (M. Duran, F. Vaz, Academic Medical Center Amsterdam) with a strongly decreased level of tetralinoleylcardiolipin was highly suggestive of the diagnosis Barth syndrome (Figure 2), which was confirmed by the mutation c.280C>T (p.Arg94Cys) in the TAZ gene (taffazin) on chromosome Xq28, which leads to an amino acid exchange and has been described before (Lekanne and Mannens, Academic Medical Center Amsterdam). The mother is not a carrier.
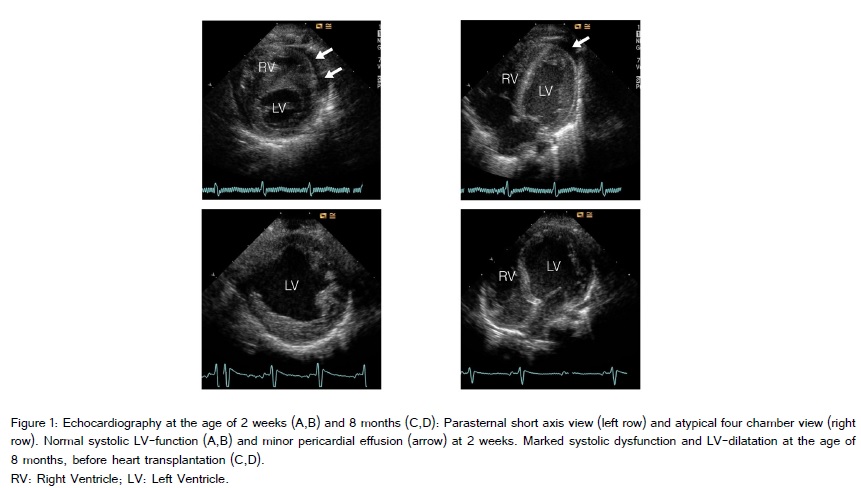
At the age of 7 months he suffered from a syncope like episode with pallor and loss of muscular tone. After this episode, for the next days he was tired and adynamic. Body weight was 5.900 g (0.5 kg below 3rd percentile), length 64 cm (3rd percentile), and the head circumference 40 cm (4 cm below 3rd percentile). Neither edemas nor murmur were present. There were good peripheral pulses and the liver was 2 cm below costal margin.
Blood chemistry showed slight elevation of liver function tests. No leukocytopenia. NT-pro-BNP was grossly elevated to 17,432 ng/L (0-65). Echocardiography showed a dilatation of left ventricle (diastolic diameter 2.8 cm) with fraction of shortening (FS) of 25%. As heart failure treatment, carvedilol 2 x 2.8 mg was started. He was breast-fed and had been started on complementary foods. At demission NT-pro-BNP was 14,000 ng/L (normal under 12 months of age up to 1,000 ng/L).
A week later he suffered from gastroenteritis with no mayor complications. However, NT-pro-BNP had risen to 31,471 ng/L, leukocyte count was 4.9 G/L, hemoglobin 10.3 T/L, lactate normal. FS was 18%. Furosemide 3 x 2 mg and spironolactone 1 x 6 mg were added to the medication.
At the age of 8 months, the boy cardially decompensated (FS 12%) (Weight 6.700 g, between 3rd and 10th percentile). To improve myocardial function, treatment with levosimendane was initiated, however function worsened, thus, a left ventricular assist-device (Berlin heart) was implanted as bridge to cardiac transplantation. He received a pediatric donor organ two weeks later. Immunosuppression was induced with ATG (antithymocyte globulin), then continued with cyclosporine A 2 x 25 mg and mycophenolate mofetil 3 x 250 mg. L-carnitine and the vitamins were discontinued.
NT-pro-BNP has stabilized around concentrations of 300ng/L. The explanted heart showed spongy degeneration of the left ventricle, compatible with LVNC.
When recovering from cardiac transplantation he showed a right-sided hemiparesis (vascular incident on left arteria cerebri media), from which he slowly recovered in the following years.
Actually the boy is ten years old, weight, height and head circumference are still under the 3rd percentile, but he is growing. Organ function is good. Immunosuppressive treatment was switched from cyclosporine A and mycophenolate mofetil to tacrolimus and mycophenolate mofetil 80 month after transplantation, due to gingiva hyperplasia and was again switched to tacrolimus and sirolimus 90 month after transplantation, due to transplant vasculopathy, detected with intravascular ultrasound (IVUS) during follow up cardiac catheterization. Repeated IVUS one year after change in immunosuppressive regimen (102 month after transplantation) showed a mild regression of transplant vasculopathy. Besides the immunosuppression and an ACE inhibitor (lisinopril) he has no additional medication.
The hemiparesis is treated with physiotherapy and a splint on the right foot. He uses his right hand less than his left hand. Growth of extremities is alike on both sides. There is a mild general muscular weakness, remarkable during effort. He is able to walk and to perform daily activities adequate to age. Cognitively he is adequate to age.
Discussion
Barth syndrome can present at a very early age and may precipitate quite quickly due to the precarious metabolic balance of the newborn. In fact, BS is related to a secondary generalized disturbance of the mitochondrial electron transfer chain, thus, both the cardiac and skeletal myopathic involvement can be explained by the dysfunction of the mitochondrial respiratory chain. In more detail, BS is related to the non-maturation of cardiolipin which causes a perturbation of the electron transfer within the respiratory chain; as the respiratory chain complexes I to V are not properly related to each other by a normal cardiolipin. In addition, the dysfunctional respiratory chain produces oxygen radicals (i.e. superoxide anions and then H2O2) in excess which are deleterious in the long-term and are related to the non-compacted cardiomyopathy [4,5].
Our patient is unique, because he survived cardiac insufficiency by bridging with an assist-device and subsequent cardiac transplantation at the early age of 8 months. The graft is working well. His cognitive development at the age of ten years is adequate. In spite of a very early diagnosis of the underlying disease, in our patient the heart insufficiency couldn?t be prevented. However, without close observation of the heart function and the possibility to bridge lack of heart function and then the option of a heart transplant, the patient would not have survived. Now a days, the bridging of the heart function can be offered to very young children and can be in place for a long time interval (i.e. 12 months) [6,7]. This is very important, as the availability of pediatric donor organs is limited. In view of these difficulties, it is essential to choose the time for intervention very carefully. In patients with underlying systemic disease besides the surveillance of heart function, the development of symptoms in other organs has to be followed carefully ? as that might be a contraindication for the option of heart transplant.
BS can present at a very early age, as early as the first day of life. Formerly, there was a high mortality during infancy and childhood, few patients survived beyond the age of four years [1]. Causes of death in BS include infections due to neutropenia and severe heart failure, which can lead to lactic acidosis and metabolic decompensation. However, one of the problems in patients with BS is that lactic and methylglutaconic acid concentrations as well as the neutropenia fluctuate [8]. On the one hand, patient with BS can have normal values for all markers in a metabolic stable period, whereas i.e. lactic acid can secondarily reach high concentrations in an agitated healthy patient ? making diagnosis difficult. Recently, easier assays for disturbances in cardiolipin metabolism have been established and the possibility for inclusion of BS in newborn screening programs is being discussed [9]. We suggest exclusion of BS by detection of abnormal cardiolipin as an early step in male patients with cardiac symptoms with or without elevations of lactic and methylglutaconic acid and neutropenia. In the same way that biochemical markers fluctuate, LVNC can be a marker of BS but is not compulsory, as our patient showed ECHOs without LVNC until shortly before the heart explantation.
With greater awareness of the disease, an increased survival of patients with Barth syndrome has been observed. Reports on transplanted children with BS are now more common [1,10-14]. Immunosuppression comprises high-dose steroids at induction of anesthesia with or without induction therapy (anti thymocyte globuline) and basiliximab, respectively. In the immediate transplant period children receive a purine synthesis inhibitor (azathioprine / mycophenolate mofetil) and a calcineurin inhibitor (tacrolimus / cyclosporine). In the post-transplant period immunosuppression usually comprises a steroid-free double therapy with a calcineurin inhibitor and a purine synthesis inhibitor. Our patient is on mycophenolate mofetil and sirolimus with good response so far. There have been no signs of acute graft rejection.
In literature, 22 patients with Barth syndrome, in which the clinical courses before one month of age have been described, show clinical courses before the age of one month ? all patients had a cardiomyopathy. However, not all report on metabolic changes, so they might have been neglected [1,2,8,10,11,14-22]. In our patient, at the age of a week, ECG showed slight ST decrement and physiological signs of right ventricular hypertrophy, on ECHO the heart looked structurally normal with good myocardial function, thus, heart function is not always affected at the early stages of Barth syndrome. However, close follow up of cardiac function is essential, as the heart is one of the target organs of the cardiolipin disturbance.
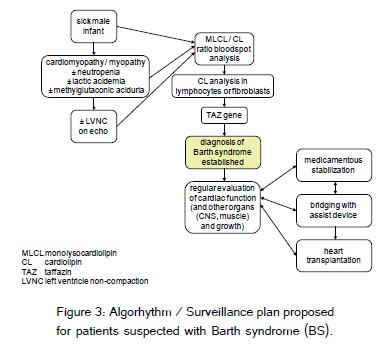
In conclusion, we emphasise that early metabolic stabilization and regular cardiac evaluation is a must for the young affected infants with BS. In this report, we suggest a surveillance plan for BS patients before heart transplantation (Figure 3).
Acknowledgement / Disclosures
We wish to thank the patient and his family for very good cooperation. There are no funding sources or conflicts of interest.